Histiocitosis de células de langerhans

La histiocitosis de células de Langerhans (LCH) es un grupo heterogéneo de enfermedades caracterizadas por la proliferación de células patológicas fenotípicamente similares a las células de Langerhans.
La histiocitosis de células de Langerhans es una enfermedad multisistémica con un curso impredecible y variable, que comprende un espectro que va desde formas que curan y otras potencialmente fatales. Estas enfermedades se conocían anteriormente como histiocitosis X, que comprendían 4 entidades bien definidas:
-Una enfermedad congénita autolimitada llamada Hashimoto-Pritzker;
-Una forma multifocal con diabetes insípida, exoftalmos, y defectos óseos llamada Hand-Schuller-Christian;
-Enfermedad Letterer-Siwe, la forma más severa de LCH, presentándose clínicamente con compromiso intertriginoso; y granuloma eosinofìlico,
-Una forma unifocal.
La histiociocitosis X se reclasificó dentro de las histiocitosis de células de Langerhans, una entidad simple con un amplio espectro clínico.
El diagnóstico se basa en las características clínicas y se confirma por la positividad de CD1a, detectado por inmunohistoquímica, o por la presencia de gránulos de Birbeck en la microscopía electrónica. El pronóstico se relaciona con la edad de inicio, la progresión de la enfermedad y la presencia de compromiso sistémico. La disfunción orgánica (hígado, pulmones, médula ósea) ocurre en el 15% de las HCL y es el factor más importante relacionado con un peor pronóstico.
Se presentó a la consulta un paciente masculino de 2 meses de edad con pápulas asintomáticas en el tronco, de 3 semanas de evolución. Al exámen físico, se observaban pápulas firmes color piel en el tronco ( fig1). El resto del exámen físico era normal. No presentaba antecedentes de deterioro del desarrollo, fiebre, ni vómitos y no presentaba linfoadenopatías palpables ni hepatoesplenomegalia.
 Figura 1. Pápulas color piel firmes en el tronco. |
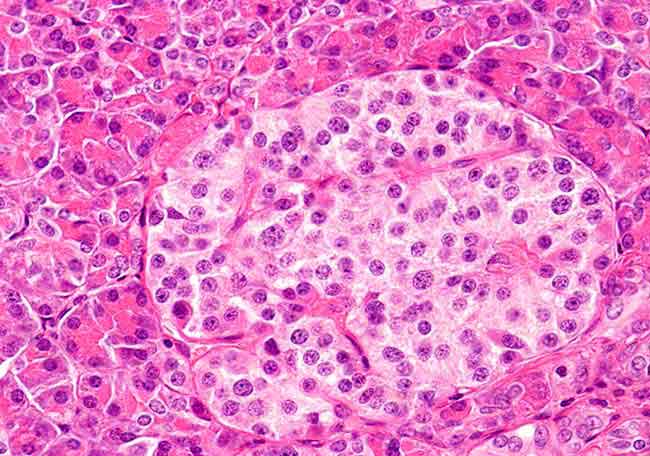
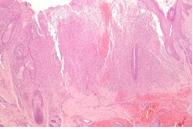 Figura 2. Infiltración de la dermis superior y epidermis con células con características de células de Langerhans (H&E, x4) |
La biopsia de piel reveló infiltración de la dermis superior y epidermis, con características de células de Langerhans (fig 2) y positividad para CD1a (fig 3) y proteina S100 (fig 4).
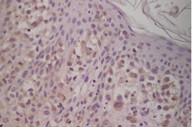 Figura 3. Positividad para CD1a (x40) |
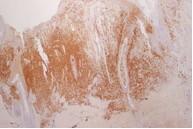 Figura 4. Positividad para proteina S100 (x4) |
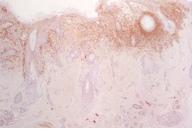
Las lesiones resolvieron espontáneamente en 2 meses, y se diagnosticó histiocitosis auto-limitada (anteriormente conocida como Hashimoto-Pritzker). Sin embargo, a los 3 meses posteriores, desarrolló eritema y maceración de ambas axilas (fig 5) y de los pliegues inguinales (fig 6). La segunda biopsia de piel mostró las mismas características que la primera, compatible con histiocitosis de células de Langerhans (figs 7, 8 y 9). Los exámenes complementarios hemograma completo, perfil químico, Rx de tórax y esqueleto no mostraron compromiso de otros órganos además de la piel. Los exámenes de laboratorio y estudios radiográficos se realizaron cada 6 meses. Actualmente, 14 meses después, el niño presenta las mismas lesiones cutáneas sin signos de progresión de la enfermedad ni de compromiso sistémico.
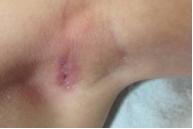 Figura 5. Eritema y maceración del pliegue axilar. |
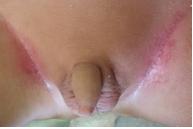 Figura 6. Eritema y maceración de los pliegues inguinales. |
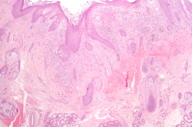 Figura 7. Pliegue axilar: Infiltración de la dermis superior y epidermis con células característica de células de Langerhans (H&E, x4) |
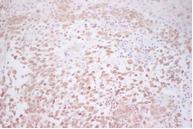 Figura 8. Pliegues axilares: Positividad para CD1a (x20) |
|
|
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.