La histiocitosis de células de Langerhans (LCH) es una enfermedad rara. Su forma de presentación varía desde lesiones simples u óseas multifocales a enfermedad ósea diseminada con compromiso multiorgánico que puede afectar la piel, bazo, pulmones, sistema nervioso central, médula ósea, tracto gastrointestinal, y nódulos linfáticos. Las lesiones que afectan diferentes órganos tienen apariencia histopatológica similar e incluyen histiocitos mezclados con un número variable de linfocitos, eosinófilos y neutrófilos. La incidencia de LCH es cerca de 2 a 5 casos por millón, con predominio masculino. Su pico de incidencia es en niños de 1-4 años, y puede ocurrir a cualquier edad.
Reporte del caso:
Un niño de 60 días fue traído al hospital con una historia de 7 días de pápulas rojizas en cuero cabelludo, tronco, extremidades, palmas y plantas. Había sido diagnosticado como eccema y tratado con corticoides en ungüento en otro hospital. Sin embargo, las pápulas persistieron y empeoraron.
Los análisis maternos fueron negativos para hepatitis C, hepatitis B, antígeno de superficie, estreptococo grupo B, HIV, clamidia, y gonorrea, pero no demostró inmunidad para el virus de la rubeola.
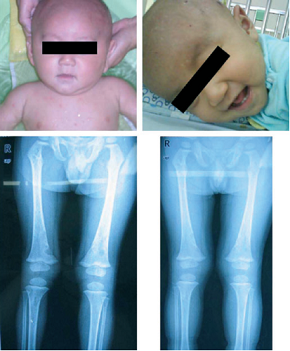
Al exámen físico, presentaba peso apropiado para la edad y estaba bien nutrido. Las pápulas estaban distribuídas en el cuero cabelludo, tronco, extremidades, palmas y plantas (fig 1 a).
Los análisis al ingreso incluyendo hemograma completo, química, análisis de orina, tests de función hepática, eran normales excepto una ligera elevación de los glóbulos blancos 10.5 x 109/l, disminución de la hemoglobina 8.2 g/l, y eritrosedimentación de 40 mm/h. Dos días después, el paciente presentó fiebre leve, con una temperatura de 37.8º C. La radiografía de tórax era normal. La tomografía computada de alta resolución (CT) mostró múltiples nódulos en el campo pulmonar derecho. Una ecografía de abdomen mostró hepatomegalia. Una radiografía simple mostró las imágenes típicas en sacabocados en la calota y lesiones líticas en fémur distal derecho (fig 1 c).
Se realizó el exámen histopatológico de una biopsia de piel de una pápula representativa. Se observaron linfocitos, eosinófilos, neutrófilos, y escasas células gigantes multinucleadas mezcladas con histiocitos. La mayoría de los histiocitos presentaban un citoplasma abundante rosado, núcleos elongados, que correspondían a histiocitos de células de Langerhans. No se observaba atipia celular ni figuras mitóticas. La tinción para histiocitos proliferantes eran positivos para S100, CD1a, CD68, y Ki 67 (-) 1%. La aspiración de médula ósea demostró una proliferación activa sin histiocitos. Se confirmó el diagnóstico de LCH. El paciente se trató con metilprednisolona (30 mg/kg IV) por 3 días, seguida de prednisona 15 mg administrada en dos dosis diarias por 4 semanas, bajándola en dos semanas y 6-mercaptopurina 20 mg diarios, metotrexate semanal 8 mg oral, y vinblastina 2.5 mg semanales administrados por inyección durante 6 semanas. La terapia continuaba con prednisona 15 mg y ciclosporina A 50 mg administrada en dos dosis por día por 5 días por 54 semanas.
Se realizaron los exámenes radiológicos (cráneo y esqueleto) y ultrasonido abdominal luego de 6, 12, y 24 semanas de tratamiento y luego de completado el tratamiento (cada 3 meses). Estos exámenes revelaron una reducción progresiva de las lesiones. Luego de 1 mes de tratamiento, la temperatura del paciente era normal. Las pápulas rojas desaparecieron a los 3 meses luego de completar la terapia, pero permanecieron hiperpigmentaciones residuales (fig 1 b).
La ecografía abdominal y CT de tórax eran normales. No se observó evidencia de enfermedad a los 9 meses de completada la terapia.
Luego de 13 meses, cuando resolvieron los signos y síntomas, la madre refirió que no podía caminar y se quejaba de dolor en la pierna derecha.
Las radiografías revelaron lesiones líticas en parte distal de fémur derecho. El paciente se internó y se trató nuevamente con quimioterapia. A las 4 semanas, las lesiones líticas de hueso en parte distal de fémur estaban en remisión (fig 1d).
A B
C D
Actualmente, el niño se siguió clínicamente por 34 meses. Se mostró mejoría de las lesiones líticas con la terapia, y la piel permaneció libre de lesiones.
La histiocitosis de células de Langerhans es una enfermedad rara de interés dermatológico. Puede presentarse simulando dermatitis crónica y debería sospecharse cuando no hay respuesta a la aplicación externa de corticoides convencionales.
La etiología de ésta enfermedad no es clara.
Clínicamente, la LCH es una enfermedad polimorfa que puede afectar uno o múltiples órganos. Es bien conocido el compromiso de hueso, piel, nódulos linfáticos, pulmones, hígado, bazo, y sistema nervioso central.
El niño presentado en éste artículo presentaba LCH multifocal y multisistémica. La histiocitosis cutánea es generalmente la manifestación inicial de la enfermedad y clásicamente se presenta como placas eritemato escamosas en cuero cabelludo, que generalmente se confunde con eccema
Las manifestaciones de LCH que afecta solo piel generalmente ocurren en niños menores de 1 año. El paciente reportado tenía 60 días de vida. Las lesiones cutáneas pueden ser la única manifestación, pero son necesarias otras investigaciones que incluyan el estudio de los huesos, para determinar si la enfermedad es más extensa.
Las lesiones óseas se encuentran comúnmente en pacientes con LCH. En los primeros 6-12 meses luego del diagnóstico, aparecen lesiones adicionales y las originales pueden progresar. Puede comprometer cualquier hueso, pero más del 50% ocurre en cráneo, pelvis, costillas y mandíbula. De los huesos largos, los más frecuentemente involucrados son las diáfisis del fémur, tibia y húmero, con endostio festoneado, adelgazamiento de la cortical, y amplitud de la cavidad medular.
La histiocitosis de células de Langerhans puede diagnosticarse en cualquier grupo etáreo pero principalmente afecta a los niños. Puede presentarse en niños en cualquier momento desde el periodo neonatal hasta los 15 años pero la incidencia es mayor entre 1-4 años. La enfermedad es rara, con una frecuencia aproximada de 2 a 5 casos por millón por año, y es ligeramente más prevalente en niños.
El pronóstico y tratamiento dependen de la extensión de la enfermedad al momento de la presentación y del compromiso de los órganos de alto riesgo como hígado, cerebro, bazo, pulmones y médula ósea.
Los niños menores de 2 años tienen peores resultados. Además de la edad de presentación, los factores pronósticos más relevantes son el compromiso multisistémico y la disfunción de órganos vitales. La linfoadenopatía y rash cutáneo se asocian con enfermedad diseminada y alto porcentaje de mortalidad.
El paciente presentado estaba en remisión total luego del diagnóstico por 1 año y posteriormente presentó recurrencia de la enfermedad en hueso. Por lo tanto, los autores consideran que la presencia de lesiones en nódulos linfáticos y en cuero cabelludo como forma de presentación de LCH indica alto riesgo de recurrencia, que es congruente con los hallazgos reportados previamente.
Las modalidades de tratamiento de LCH varían dependiendo de la localización de la enfermedad, extensión, y del número de lesiones. En casos de disfunción orgánica, está indicada la quimioterapia sistémica. El objetivo del tratamiento es incrementar la sobrevida y reducir la incidencia de secuelas permanentes acelerando la regresión de infiltrados. Las lesiones óseas extensas o múltiples y la enfermedad multisistémica se tratan con terapia inmunomoduladora, que incluye glucocorticoides sistémicos (prednisona), quimioterapia (vinblastina), y antagonistas de los folatos (metotrexato), y análogos de las purinas (6-mercaptopurina).
En éste paciente, se administró quimioterapia por el compromiso multifocal y multisistémico de la enfermedad. Los porcentajes de recurrencia dependen de la estrategia del tratamiento y localización de las lesiones y se reporta que varían desde el 1.6 al 25%. Los pacientes deberían seguirse por un largo período.
En conclusión, los autores presentaron un paciente con LCH. Se destaca la necesidad de administrar regímenes de tratamiento más largos que pueden reducir el porcentaje de recurrencia en casos de compromiso multifocal y multisistémico. Aunque el paciente mostró alguna evidencia de recurrencia, es posible que una variedad de modalidades de tratamiento y el seguimiento cuidadoso pueda lograr resultados satisfactorios.
¿Qué aporta éste artículo a la práctica dermatológica?.
Aunque es muy rara, la LCH puede ser amenazante para la vida. El pronóstico depende de la edad del paciente, la extensión de la enfermedad y de la afectación de órganos vitales. En casos de disfunción de órganos, está indicada la quimioterapia sistémica. El diagnóstico temprano puede mejorar la sobrevida. Estos datos muestran la importancia de conocer mejor la enfermedad, para diagnosticarla tempranamente y disminuir la probabilidad de secuelas. Se deberían realizar estudios prospectivos para conocer sobre la LCH y mejorar el manejo clínico de niños con ésta enfermedad.
♦Comentario y resúmen objetivo: Dra. Geraldina Rodriguez Rivello