Espectro de la neuromielitis óptica
| Introducción |
El trastorno del espectro de la neuromielitis óptica (NMOSD, por sus siglas en inglés) es una enfermedad autoinmune rara. Provoca una inflamación multifocal del sistema nervioso central (SNC) que afecta principalmente a los nervios ópticos y la médula espinal, y típicamente resulta en ataques de pérdida visual y parálisis.
Las asociaciones entre la enfermedad del nervio óptico y de la médula espinal se observaron en el siglo XIX, especialmente después de la descripción de Devic de “neuro-myélite optique aiguë” (neuromielitis óptica aguda).1
En el siglo XX, la “enfermedad de Devic” se consideraba una variante óptica monofásica y espinal grave de la esclerosis múltiple que no comprometió el cerebro.2 Sin embargo, el descubrimiento de los anticuerpos de clase IgG que se unen al canal de agua acuaporina-4 (AQP4-IgG) en suero de pacientes con enfermedad de Devic, pero no de aquellos con esclerosis múltiple típica, estableció la neuromielitis óptica como una entidad distinta con un curso crónico y recidivante.3-5
Los estudios de pacientes que son AQP4-IgG-seropositivos han llevado al reconocimiento de que se producen muchos hallazgos clínicos y de neuroimagen adicionales, incluida la afectación cerebral, que se incluyen con el término trastorno del espectro de la neuromielitis óptica.6,7 El autoanticuerpo AQP4-IgG es detectable en más del 80% de los pacientes con NMOSD y es patogénico, iniciando las lesiones inflamatorias del SNC y las manifestaciones clínicas de la enfermedad.8,9
| Epidemiología |
NMOSD representa del 1 al 2% de todos los casos de enfermedad desmielinizante inflamatoria del SNC en los Estados Unidos y Europa, siendo la esclerosis múltiple mucho más común, pero NMOSD representa un tercio o más de los casos de inflamación del SNC en poblaciones asiáticas y otras poblaciones no blancas.
Las estimaciones de incidencia y prevalencia oscilaron entre 0,037 y 0,73 casos por 100.000 personas-año y entre 0,7 y 10,0 casos por 100.000 personas, respectivamente, pero estas estimaciones varían ampliamente entre los estudios, con las tasas más altas entre los africanos y en la región afrocaribeña y las tasas más bajas entre los blancos y las personas en Australasia, lo que probablemente refleja las influencias genéticas y ambientales y las diferencias en los métodos de verificación.10
Un estudio basado en la población en los Estados Unidos mostró una mayor prevalencia para los negros (13,0 casos por 100.000 personas) que para los blancos (4,0 por 100.000).11
La mediana de edad al inicio del trastorno es de 40 años, pero el trastorno puede afectar personas de cualquier edad, y hasta un 20% de los casos son en niños o en adultos mayores de 65 años.
La enfermedad seropositiva tiene una preponderancia femenina que se acerca al 90%, mientras que los casos seronegativos tienen una distribución igual por sexos.10,12 Hasta un 3% de los casos son familiares.13 La secuenciación del genoma completo ha demostrado que AQP4-IgG se asocia con variantes de la región del complejo mayor de histocompatibilidad, y existe una relación causal entre las variantes de riesgo conocidas y el lupus eritematoso sistémico, que puede coexistir con NMOSD positivo para anticuerpos.14
Se ha sugerido que los factores ambientales pueden desempeñar un papel en la génesis del trastorno, pero no se ha identificado ninguno. En NMOSD pediátrico, a diferencia de la esclerosis múltiple, las exposiciones comunes al virus herpes, incluido el virus de Epstein-Barr, no están implicadas.15
Los ataques agudos de NMOSD, incluido el episodio inicial, generalmente ocurren en ausencia de un evento desencadenante identificado, pero aproximadamente un tercio de los casos son precedidos por una infección, que es a menudo viral, y en casos raros, un ataque agudo sigue a la vacunación.2 Sin embargo, ninguna infección específica o vacuna ha sido fuertemente implicada como desencadenante de la actividad de la enfermedad. El ataque inicial de NMOSD con AQP4-IgG se ha asociado con el uso de las terapias con inhibidores de puntos de control inmunitarios.16,17
| Características clínicas |
Se han identificado seis síndromes clínicos centrales en NMOSD.
Se clasifican por su ubicación: nervio óptico, médula espinal, área postrema de la médula dorsal, otras regiones del tronco encefálico, diencéfalo o cerebro. Estos síndromes se manifiestan como ataques agudos y recaídas de disfunción neurológica, con síntomas que típicamente evolucionan durante un período de días.7
Los síndromes más comunes son la neuritis óptica y la mielitis transversa, así como el síndrome del área postrema, que provoca vómitos intratables o repetitivos e hipo. Las lesiones multifocales pueden dar lugar a una combinación simultánea o rápidamente secuencial de síndromes (p. ej., neuritis óptica y mielitis).
Los ataques se identifican sobre la base de nuevos síntomas clínicos y signos neurológicos objetivos (excepto en el caso de síntomas aislados del área postrema) y descartando otras causas, como infección, que puede simular un ataque. La detección de nuevas lesiones en imágenes de resonancia magnética (RM) en una región neuroanatómica relevante proporciona evidencia de apoyo de que un evento neurológico se debe a NMOSD.
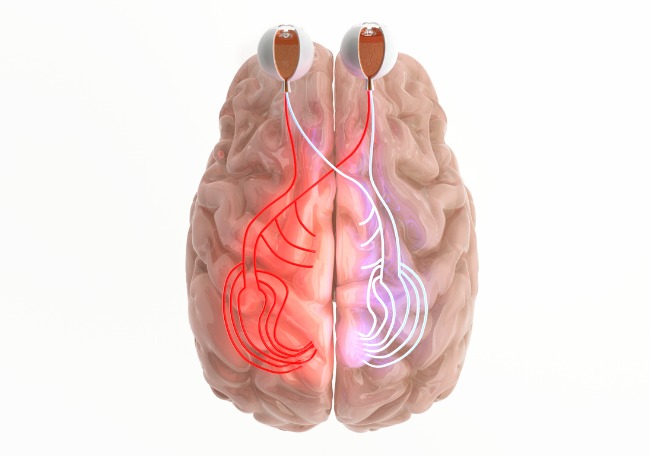
El ataque centinela involucra el nervio óptico o médula espinal en más del 85% de los adultos afectados. Los pacientes con neuritis óptica se presentan con pérdida visual unilateral o bilateral o escotoma, discromatopsia y dolor ocular exacerbado por el movimiento del ojo que no se distingue de la neuritis óptica en la esclerosis múltiple o de una forma idiopática.
La mielitis transversa aguda causa debilidad en las extremidades, entumecimiento, pérdida sensorial o dolor debajo del nivel de la lesión y disfunción vesical e intestinal. Otros síntomas de mielitis incluyen el signo de Lhermitte (parestesias del tronco o de las extremidades), provocadas por la flexión del cuello y episodios de espasmos tónicos paroxísticos que duran de 15 a 60 segundos, con contracción muscular agonista y antagonista dolorosa, a menudo confundida con convulsiones. La mielitis cervical alta severa puede causar insuficiencia respiratoria neurogénica.2
El síndrome del área postrema es la forma de presentación característica de NMOSD en el 10% de los pacientes y ocurre en algún momento durante la enfermedad en el 15 al 40% de los pacientes.18,19 Los vómitos duran una mediana de 2 semanas y, hasta en dos tercios de los casos, presagian un ataque de neuritis o mielitis óptica.19
El síndrome puede acompañar a la mielitis cuando una lesión cervical destructiva asciende al tronco encefálico, o puede ser aislado y causado por lesiones medulares dorsales no destructivas. Cuando el síndrome es aislado, especialmente como la presentación inaugural de NMOSD, los pacientes son típicamente evaluados por internistas o gastroenterólogos por vómitos intratables, y el diagnóstico puede retrasarse hasta que haya síntomas neurológicos posteriores.
Los otros síndromes NMOSD ocurren con menos frecuencia, excepto que los síndromes cerebrales son comunes en los niños y causan signos generalizados o focales, incluyendo encefalopatía, hemiparesia, hemianopsia o convulsiones.7,20
Las lesiones del tronco encefálico pueden causar disfunción oculomotora, pérdida de audición, vértigo, disartria u otros síntomas de los nervios craneales; al igual que con la neuritis óptica, estas características por sí solas no son inicialmente distinguibles de síndromes similares en la esclerosis múltiple. Se informó que lesiones en estructuras diencefálicas como el tálamo y el hipotálamo causan síntomas similares a la narcolepsia, endocrinopatías, desregulación de la temperatura, trastornos alimentarios y un síndrome de secreción inadecuada de hormona antidiurética.
| Neuroimagen |
La confirmación de que un ataque se debe a NMOSD es ayudada en gran medida por la detección en la RM de lesiones características en el cerebro, el nervio óptico, el quiasma óptico o la médula espinal.21 En la fase aguda, las secuencias de RM ponderadas en T2 pueden mostrar lesiones inflamatorias nuevas o agrandamiento de las lesiones, y secuencias ponderadas en T1 obtenidas con gadolinio pueden mostrar realce de lesiones activamente inflamadas.
Las anomalías en la RM orbitaria tienen predilección por el quiasma óptico o el nervio óptico posterior adyacente, pero pueden ocupar toda la longitud del nervio.
La resonancia magnética de la médula espinal típicamente muestra longitudinalmente mielitis transversa extensa, definida como una lesión que es al menos tres segmentos vertebrales contiguos de longitud.2,22 Aunque este patrón es característico de NMOSD, alrededor del 15% de los ataques centinela de mielitis se asocian con una lesión más corta que simula la esclerosis múltiple.23
La detección de una lesión medular dorsal con el uso de RM confirma el síndrome del área postrema, pero la lesión es pequeña y visible por pocos días.7 Vistas en RMs, las lesiones focales en el tronco encefálico, diencéfalo o cerebro, incluso aquellas que son asintomáticas, también puede ser indicativas de NMOSD.7
| Diagnóstico |
Los criterios basados en consensos internacionales para el diagnóstico de NMOSD7 requieren la manifestación de uno o más de los seis síndromes típicos y enfatizan que las correlaciones clínico-radiológicas y una prueba serológica positiva para AQP4-IgG distingue el NMOSD de la esclerosis múltiple y otros trastornos.
Alrededor del 80% de los casos son seropositivos, pero los criterios actuales permiten el diagnóstico de NMOSD si la prueba AQP4-IgG es negativa o no disponible, que puede ser el caso en algunas regiones de bajos ingresos del mundo. Sin embargo, según los criterios de consenso internacional, el diagnóstico de NMOSD está casi asegurado si hay un primer ataque característico y los anticuerpos AQP4-IgG están presentes.7
La prueba de referencia estándar de suero AQP4-IgG es un ensayo de citometría de flujo basado en células vivas con más del 80% de sensibilidad y más del 99% especificidad.24,25 Pueden ocurrir resultados de títulos bajos falsos positivos utilizando ensayos inmunoabsorbentes ligados a enzimas y, en un caso atípico, debe impulsar la reconsideración del diagnóstico y la confirmación con un ensayo basado en células.
Los resultados falsos negativos de AQP4-IgG pueden ocurrir si el suero es muestreado durante el tratamiento con fármacos inmunosupresores o después del intercambio de plasma. Se recomienda una nueva prueba en estos casos, con un intervalo después de la interrupción del tratamiento o durante una recaída.7 La prueba de líquido cefalorraquídeo AQP4-IgG es relativamente insensible.
La categorización de NMOSD con AQP4-IgG negativo o desconocido requiere criterios clínicos y de RM y una búsqueda de otras causas de los síntomas del SNC.7 El diagnóstico alternativo más común es un trastorno desmielinizante del SNC asociado con anticuerpos séricos contra la glicoproteína de oligodendrocitos de la mielina (MOG-IgG). Este trastorno se superpone tanto con el NMOSD seropositivo para AQP4-IgG como con la esclerosis múltiple, los cuales también pueden causar neuritis óptica o mielitis.26
Los pacientes con síndromes clínicos de NMOSD generalmente se someten a pruebas para ambos anticuerpos; sin embargo, los anticuerpos AQP4-IgG y MOG-IgG rara vez coexisten. Pacientes que dan negativo para ambos anticuerpos pueden describirse como NMOSD "doble seronegativo", una categoría que podría reducirse con el descubrimiento de nuevos autoanticuerpos.
| Curso de la enfermedad |
Los ataques de NMOSD generalmente alcanzan la severidad máxima en varios días, se estabilizan y luego desaparecen espontáneamente, dejando con frecuencia déficits funcionales de moderados a severos y permanentes.27 En más del 90% de los casos, la enfermedad tiene un curso recurrente, similar a la de la esclerosis múltiple, con un período de meses o años entre ataques.
El deterioro neurológico es estable o puede disminuir durante la remisión, pero las recaídas conducen a la acumulación gradual de discapacidad. El tipo, la frecuencia y la gravedad de las recaídas están influenciadas por la edad, el sexo y la etnia. 28,29 Los datos sobre estos factores han sido obtenidos de series retrospectivas. Entre mujeres con NMOSD seropositivos, las tasas de recaída han sido mayores durante los primeros 3 meses después del parto que en el período anterior o durante el embarazo, y el riesgo de aborto espontáneo se ha incrementado.30
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.