El síndrome urémico hemolítico (SUH) se caracteriza por la tríada de anemia hemolítica microangiopática, trombocitopenia, y lesión renal aguda (LRA). Aproximadamente el 90% de los pacientes pediátricos desarrollan este síndrome después de la infección por Shigella dysenteriae (S. dysenteriae), que produce verdaderas toxinas Shiga, o Escherichia coli (E. coli), con algunas cepas produciendo toxinas simil Shiga (Shiga-like).
La toxina Shiga fue denominada originalmente Verotoxina porque las células Vero derivadas de las células epiteliales de riñón de mono verde africano eran hipersensibles a esta toxina. Posteriormente, otras toxinas fueron llamadas toxinas Shiga-like debido a sus similitudes con la toxina Shiga en términos de antigenicidad y estructura. La toxina Shiga like-1 se diferencia de la toxina Shiga por un sólo aminoácido, mientras que la toxina Shiga like-2 comparte un 56% de homología de secuencia con la toxina Shiga like-1.
Aunque las cepas de E. coli productoras de toxina Shiga-like pueden desencadenar más a menudo SUH (SUH-ECTS), ciertas cepas de S. dysenteriae secretoras de toxina Shiga también pueden causar síndrome urémico hemolítico. En la actualidad se las conoce como familia de toxinas Shiga, y los términos se usan indistintamente. El SUH producido a partir de la infección por SUH-ECST fue denominado formalmente diarrea + SUH (D + SUH) o SUH típico.
Por el contrario, el SUH que no está relacionado a las toxinas Shiga y representa aproximadamente el 10% de todos los casos de SUH, se llama síndrome urémico hemolítico atípico (SUHa). Aunque el SUH-ECTS es relativamente común en los niños, el SUHa ocurre en individuos de todas las edades y a menudo es familiar. El pronóstico es muy pobre; el primer episodio de SUHa está asociado con una tasa de mortalidad de aproximadamente 25%, y aproximadamente el 50% de los casos resultan en enfermedad renal terminal que requiere diálisis.
En los últimos años, las alteraciones en los mecanismos subyacentes de la regulación del complemento han sido focalizadas como causas de SUHa. Varias anomalías genéticas en los factores reguladores del complemento, incluyendo el factor H del complemento, se han observado en el 50-60% de los pacientes. El análisis de la patología subyacente de esta condición está progresando rápidamente.
El diagnóstico diferencial del SUHa con respecto al SUH-ECST o la púrpura trombocitopénica trombótica (PTT), otra forma de microangiopatía trombótica (MAT) causada por una deficiencia de ADAMTS13 (una desintegrina y metaloproteinasa con una trombospondina tipo 1, patrón 13), no es necesariamente fácil en las primeras etapas de la aparición de la enfermedad.
Sin embargo, si se retrasa el tratamiento, hay un alto riesgo de que este síndrome progrese a insuficiencia renal. Por lo tanto, el Comité Conjunto de la Sociedad Japonesa de Nefrología y la Sociedad Pediátrica de Japón (Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society (JSN/JPS)) han desarrollado criterios diagnósticos para el SUHa a fin de permitir su diagnóstico precoz y facilitar el inicio oportuno del tratamiento apropiado.
Los autores esperan que los criterios diagnósticos presentados en este informe lleguen a tantos médicos como sea posible y sean ampliamente utilizados.
Definición de SUHa
El SUHa es un tipo de MAT que difiere del SUH-ECST y de la PTT, siendo esta última causada por una marcada reducción de la actividad de ADAMTS13. El SUHa es un síndrome caracterizado por la tríada de anemia hemolítica microangiopática, trombocitopenia, y LRA, que es similar al ECST-SUH.
Guías para el diagnóstico de SUHa
Diagnóstico definitivo
El diagnóstico definitivo de SUHa se hace cuando está presente la tríada de anemia hemolítica microangiopática, trombocitopenia y LRA. La enfermedad no debe asociarse con toxinas Shiga y también debe ser excluida la PTT.
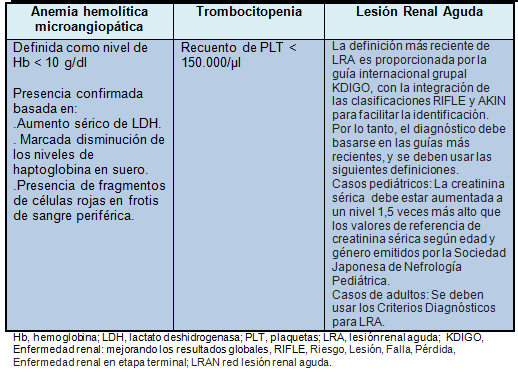
El Comité Conjunto JSN/JPS definió a la anemia hemolítica microangiopática en base a un nivel de hemoglobina (Hb) <10 g/dl. La presencia de anemia hemolítica microangiopática debe ser confirmada en base al aumento de los niveles de lactato sérico deshidrogenasa, una marcada disminución en los niveles de haptoglobina en suero, y la presencia de fragmentos de células rojas de la sangre en un frotis de sangre periférica.
La trombocitopenia se definió como un recuento de plaquetas (PLT) <150.000/µl.
La definición de LRA ha sido actualizada, con la definición más reciente propuesta por la guía internacional grupal “Enfermedad Renal: Mejorando los Resultados Globales” que integra las clasificaciones de Riesgo, Lesión, Falla, Pérdida, Enfermedad renal en etapa terminal y las clasificaciones del Grupo de Trabajo sobre Lesión Renal Aguda para facilitar su identificación.
Por lo tanto, los autores recomiendan el diagnóstico basado en las guías más recientes, junto con las siguientes definiciones. Para los casos pediátricos, la creatinina sérica debe incrementarse a un nivel 1,5 veces mayor que los valores de referencia de creatinina sérica según edad y sexo emitidos por la Sociedad Japonesa de Nefrología Pediátrica. Para los casos de adultos, se deben utilizar los criterios diagnósticos para LRA.
Pautas para el diagnóstico de SUHa
Diagnóstico definitivo
El diagnóstico definitivo de SUHa se hace cuando está presente la tríada de anemia hemolítica microangiopática, trombocitopenia y LRA. La enfermedad no debe estar asociada con las toxinas Shiga y también debe ser excluida la PTT. La tabla 1 presenta las definiciones de anemia hemolítica microangiopática, trombocitopenia y LRA establecidas por el Comité Conjunto JSN/JPS.
Tabla 1. Definiciones de anemia hemolítica microangiopática, trombocitopenia y LRA establecidos por el comité conjunto de la JSN/JPS
Diagnóstico probable
Un diagnóstico probable de SUHa se hace cuando se encuentran 2 de las 3 siguientes condiciones: anemia hemolítica microangiopática, trombocitopenia y LRA. La enfermedad no debe tener ninguna asociación con toxinas Shiga y debe ser excluida la PTT.
Aplicación de los criterios diagnósticos
Cuando los autores aplicaron estos criterios diagnósticos en la cohorte TMA de la Universidad de Medicina de Nara (UMN), 15 de los 37 individuos que tenían todos los datos necesarios para la evaluación fueron diagnosticados con SUHa definitivo. Dado que los datos se registraron en un solo punto de tiempo, se especuló que la sensibilidad de los criterios diagnósticos aumentaría si se pudieran evaluar los datos desde múltiples puntos de tiempo.
El valor de corte para la anemia, que se definió como un nivel de Hb <10 g/dl, y el valor de corte para la trombocitopenia, que se definió como un recuento de PLT <150.000 µ/l, son equivalentes a los utilizados por el Registro Internacional de SUH/PTT recurrente y familiar.
Los autores consideraron el uso de un valor de corte de PLT <100.000 µ/l para la trombocitopenia para reflejar aquel utilizado en los criterios diagnósticos de SUH-STEC de la Sociedad Japonesa de Nefrología Pediátrica (2000), pero sólo hallaron 1 paciente con un recuento de PLT entre 100.000-150.000 µ/l en la cohorte de la UMN. Por lo tanto, es probable que esta diferencia no tuviera un gran impacto en la sensibilidad o especificidad de los criterios diagnósticos.
Los criterios diagnósticos propuestos por los autores incluyen la categoría de SUHa "Probable" porque consideran que este diagnóstico tentativo ayudará en el diagnóstico precoz del SUHa y podría evitar retrasos en el desarrollo de enfoques terapéuticos apropiados para los pacientes con esta patología.
Evaluación de la activación inapropiada del complemento
Las anomalías en la regulación del complemento se encuentran entre las principales causas de SUHa. El diagnóstico de SUHa causado por la activación inapropiada del complemento se ha vuelto más crítico debido al eculizumab, un anticuerpo monoclonal anti-C5 humanizado, que ha demostrado ser una modalidad terapéutica eficaz y ha sido aprobado para el tratamiento de pacientes con SUHa en Europa y los Estados Unidos.
Recientemente, Fan y colegas evaluaron las relaciones genotipo-fenotipo en pacientes japoneses con SUHa e identificaron mutaciones potencialmente causales en el factor H del complemento, C3, proteína cofactor de membrana, y trombomodulina en 8 de los pacientes.
Sin embargo, el diagnóstico definitivo de la activación inapropiada del complemento en pacientes con SUHa es difícil debido a que algunos de ellos muestran niveles séricos normales de los componentes del complemento y a que existe una serie de proteínas reguladoras del complemento, por lo que es difícil decidir cual proteína reguladora del complemento es responsable del desarrollo de SUHa en un paciente en particular.
Exclusión de la infección por E. coli productora de toxina Shiga
El SUH-ECST se caracteriza por diarrea acompañada de sangre en las heces. Sin embargo, la diarrea también puede estar presente en algunos casos de SUHa. La diarrea en el SUHa puede ser una manifestación de colitis isquémica. Además, la enteritis que no es causada por ECST puede desencadenar SUHa. Por lo tanto, el diagnóstico de SUH-ECST no puede hacerse basado en los síntomas, y la nomenclatura anterior que utilizaba "SUH D +" para corresponderse con el SUH-ECST y "SUH D -" para el SUHa no se utiliza en la actualidad. La participación de las toxinas Shiga debe confirmarse mediante cultivo de heces, la detección directa de toxinas Shiga, o la detección de anticuerpos IgM anti-lipopolisacárido.
Exclusión de la PTT
Convencionalmente, la PTT se ha diagnosticado en base a la péntada clásica (anemia hemolítica microangiopática, trombocitopenia, trastorno psiconeurótico lábil, fiebre y falla renal).
Sin embargo, el descubrimiento del ADAMTS 13 ha llevado a la conclusión de que el 60-90% de los pacientes con PTT tiene una marcada reducción en la actividad de ADAMTS 13, a un nivel <5%, independientemente de la raza. Por lo tanto, cuando se diagnostica SUHa, los pacientes que tienen disminución marcada de los niveles de actividad de ADAMTS 13 (<5%) deben ser diagnosticados como PTT, lo que excluye el diagnóstico de SUH. Sin embargo, algunos pacientes pueden mostrar la péntada clásica de PTT y tener niveles normales o ligeramente reducidos de actividad ADAMTS 13.
Por lo tanto, si un paciente tiene niveles de actividad ADAMTS13 ≥ 5%, para el diagnóstico diferencial de SUHa o PTT puede ser necesario tener en cuenta otros síntomas clínicos.
Exclusión de MAT causada por otros factores
Las enfermedades que evidentemente causan un estado clínico de MAT, incluyendo coagulación intravascular diseminada, riñón esclerodermatoso e hipertensión maligna, se deben excluir cuando se diagnostica SUHa.
Cuando se sospecha un caso probable de SUHa
Cuando se sospecha un caso probable de SUHa, se deben recoger todas las muestras que sean necesarias para determinar el diagnóstico adecuado, y la estrategia terapéutica debería establecerse previa consulta con una institución que cuente con una amplia experiencia en el manejo de casos de SUHa.
Casos en los que se debe sospechar fuertemente SUHa
Si existen rasgos que son característicos del síndrome urémico hemolítico, debe sospecharse fuertemente el SUHa si se cumplen los siguientes requisitos, independientemente de la presencia de diarrea: si el paciente es menor de 6 meses de edad; si el tiempo de aparición no está claro (aparición latente); si el paciente tiene antecedentes de síndrome urémico hemolítico (caso recurrente); si el paciente tiene antecedentes de anemia de causa desconocida; si hay SUH recurrente después de un trasplante de riñón; si el paciente tiene antecedentes familiares de síndrome urémico hemolítico (excepto los casos de intoxicación por alimentos); y si el paciente no tiene diarrea o heces con sangre.
Clasificación de causas de SUHa, con exclusión de la PTT causada por defecto de ADAMTS 13
La Tabla 2 clasifica las causas de SUHa y presenta los métodos para determinarlas.