| INTRODUCCIÓN |
Los tumores oftálmicos pediátricos se presentan al nacer o se adquieren durante la infancia y son de etiología variada. Aunque la mayoría son benignos, los tumores malignos pueden ser potencialmente mortales y es imperativo un diagnóstico precoz. Tanto las lesiones benignas como las malignas pueden causar pérdida de la visión debido a su efecto sobre las estructuras oculares vitales, así como a factores ambliogénicos, que deben tenerse en cuenta en niños pequeños cuyo sistema visual está en desarrollo. Ciertos tumores, en particular los de origen vascular y neurogénico, pueden estar asociados con enfermedad sistémica, y puede estar indicada una evaluación diagnóstica adicional. El pediatra es a menudo el primero en encontrar tumores oftálmicos y debe estar familiarizado con los diagnósticos y presentaciones comunes de la enfermedad.
> Presentación de los tumores oftálmicos
La presentación depende de la ubicación de la lesión. Las lesiones externas del párpado y de la superficie del ojo son típicamente evidentes, mientras que las lesiones intraoculares y orbitarias no se pueden visualizar solo con una inspección macroscópica. En cambio, el paciente presentará varios signos que pueden alertar al médico de una patología ocular. Aunque la disminución de la visión puede ser un síntoma de presentación en todos los tumores oftálmicos, es importante señalar que los niños a menudo no se quejan de mala visión, especialmente si el proceso es unilateral.
> Examen
El examen comienza con la evaluación de la agudeza visual de cada ojo, adaptada a la edad y el estado de desarrollo del paciente; evaluación de las pupilas para determinar simetría en tamaño y reflejo rojo, reactividad a la luz y defecto pupilar aferente; y examen de la motilidad, evaluación de los movimientos extraoculares y evaluación de estrabismo. Se debe inspeccionar toda la cara para detectar asimetría o anomalía, seguido de un examen externo del ojo y del fondo de ojo. El fotocribado es una herramienta complementaria útil para evaluar el error refractivo, la leucocoria y el estrabismo. Los detalles del examen ocular pediátrico se pueden encontrar en referencias externas.
| TUMORES DEL PÁRPADO |
> Presentación
Las lesiones del párpado se presentan como protuberancias o nódulos en el párpado. Los tumores grandes del párpado pueden causar ptosis que obstruye el eje visual, y el efecto de masa en el globo puede inducir astigmatismo, lo que conduce a una posible privación o ambliopía anisometrópica (enfoque desigual entre los 2 ojos que causa desenfoque crónico en 1 retina), respectivamente.
> Examen
Los tumores de párpados se diagnostican a menudo en base únicamente a la historia y la visualización. Se debe prestar atención al margen del párpado en caso de conjuntivitis porque lesiones pequeñas y poco visibles pueden ser las responsables. La pérdida focal de pestañas a nivel del tumor puede ser un signo de malignidad, lo cual es poco común en la población pediátrica. Aunque a menudo no es necesario realizar más pruebas con imágenes, puede ser necesaria una biopsia cuando se requiere un diagnóstico definitivo. A veces, las lesiones orbitarias más profundas pueden visualizarse como una lesión del párpado, representando la punta del iceberg.
> Lesiones que simulan tumores de párpado
Los tumores de párpado simulados son, con diferencia, el tipo más común de lesión de párpado y se encuentran con regularidad en la práctica.
Chalazión. Los chalaziones, las lesiones más comunes de los párpados, son causados por el bloqueo de las glándulas sebáceas de los párpados y se presentan como nódulos indoloros de crecimiento lento de tamaño subcentimétrico.(6) La mayoría de los chalaziones se resuelven con compresas tibias, masaje del párpado e higiene. Las lesiones persistentes pueden requerir tratamiento adicional. Las opciones incluyen macrólidos orales o doxiciclina en niños mayores de 8 años, inyección de corticoesteroides y escisión quirúrgica.
Orzuelo. Los orzuelos son infecciones bacterianas agudas del párpado causadas por el bloqueo de las glándulas apocrinas o ecrinas. La blefaritis, la dermatitis seborreica y la rosácea son factores de riesgo. Los orzuelos, que se presentan como pústulas dolorosas a lo largo de la línea de las pestañas, son autolimitados y duran de 1 a 2 semanas. Las compresas tibias y el masaje del párpado son la terapia de primera línea. También se utilizan antibióticos tópicos o combinaciones de antibióticos y corticoesteroides. El orzuelo puede provocar celulitis preseptal.
Molusco contagioso. El molusco contagioso es una infección benigna y autolimitada causada por un poxvirus. Se propaga mediante transmisión directa y es más común en niños de 2 a 5 años. Se presenta como pápulas perladas en forma de cúpula con umbilicación central. Generalmente asintomáticas, las pápulas pueden causar irritación o conjuntivitis secundaria. El molusco contagioso se resuelve espontáneamente; sin embargo, esto puede tardar 6 meses o más. Si hay complicaciones secundarias como conjuntivitis, se recomienda tratamiento. Las terapias incluyen agentes antivirales tópicos, escisión quirúrgica, crioterapia y cauterización.
Hemangioma en vino de Oporto (nevo flamígero facial). Los hemangiomas en vino de Oporto son malformaciones capilares ubicadas en la piel. Se presentan al nacer como lesiones planas y rojas y crecen con el niño. Pueden volverse irregulares y nodulares con la edad. Los hemangiomas en vino de Oporto faciales, en particular aquellos con una distribución que corresponde al desarrollo de la vasculatura embrionaria, se asocian con el síndrome de Sturge-Weber, una facomatosis que involucra hemangiomas en piel, cerebro y ojos. Los hemangiomas en vino de Oporto del párpado superior, especialmente en el síndrome de Sturge-Weber, pueden asociarse con glaucoma congénito y juvenil.
Nevos melanocíticos. Los nevos palpebrales son acumulaciones benignas de melanocitos con pigmentación variable y son idénticos a los que aparecen en otras áreas de la piel. Los nevos de unión y compuestos se adquieren durante la pubertad.
Los nevos azules se presentan como nódulos bien delimitados de color azul a negro y suelen ser adquiridos y solitarios por naturaleza; los que son congénitos y se distribuyen de forma difusa pueden estar asociados con síndromes familiares, como LAMB (lentigos, mixomas auriculares y nevos azules) y NAME (nevos, mixoma auricular, neurofibromas mixoides y efélides).
Los nevos en beso, un fenómeno embriológico poco frecuente, se producen en los párpados superior e inferior opuestos, llamados así por su apariencia de “beso” cuando el ojo está cerrado. Si los nevos en beso son grandes y bloquean el eje visual, se requiere intervención en la infancia.
Los nevos pilosos gigantes congénitos se presentan al nacer o en las primeras semanas posteriores al nacimiento. Pueden tener un crecimiento excesivo de pelo y alcanzar un diámetro mayor de 20 cm, pero la afectación de la órbita es extremadamente rara.
> Cáncer maligno de párpado
Aunque son poco frecuentes en niños, pueden ocurrir cánceres de piel periorbitales como el carcinoma basocelular. Por lo general, se presentan afecciones sistémicas asociadas (Tabla 1). Las lesiones premalignas, como el nevo piloso gigante y la melanocitosis ocular, predisponen a los pacientes al melanoma. También pueden aparecer en el párpado infiltrados leucémicos/linfomatosos, sarcoma de Kaposi e histiocitosis (como la histiocitosis de células de Langerhans). Se requiere una biopsia de tejido para el diagnóstico y el tratamiento es generalmente quirúrgico. Dada la ubicación, se puede realizar una cirugía de Mohs (extirpación de capas delgadas de tejido hasta que no quede cáncer) y es imprescindible una reconstrucción cuidadosa para preservar la arquitectura y la función del párpado.
| TUMORES DE LA SUPERFICIE OCULAR |
> Presentación
Los tumores de la superficie ocular incluyen los de la córnea, la conjuntiva y la esclerótica y se visualizan en un examen externo. Los pacientes presentan un bulto, pigmentación anormal o enrojecimiento de la superficie del ojo. Pueden ser sintomáticos y causar irritación o molestias oculares.
> Examen
El examen externo proporciona detalles de la superficie ocular, incluida la ubicación, la pigmentación y el tamaño del tumor, y se realiza fácilmente con una linterna en el consultorio del pediatra. Si está disponible, una lámpara de hendidura portátil es útil para la ampliación. Las lesiones pueden ocultarse en los fondos de saco y pueden no ser evidentes sin la retracción del párpado. Se deben obtener antecedentes sobre cronicidad, crecimiento y cambio de color. El examen clínico suele ser suficiente para el diagnóstico.
> Coristomas epibulbares
Los coristomas son un crecimiento excesivo de tejido normal en una ubicación aberrante y son el tumor de la superficie ocular más común presente al nacer. Pueden ser simples, conteniendo 1 elemento tisular, o complejos, conteniendo más de 1 tipo de tejido. Aparecen como una masa opaca, de color blanco amarillento en la superficie ocular. Generalmente asintomáticos, los coristomas epibulbares (en la superficie del globo ocular) pueden bloquear directamente el eje visual si se encuentran en la córnea o causar astigmatismo. Las lesiones ambliogénicas deben extirparse, mientras que las que no afectan la visión pueden observarse. Los dermoides conjuntivales se localizan por lo general en el limbo y se asocian con el síndrome de Goldenhar. Los coristomas del complejo epibulbar se observan en el síndrome del nevo sebáceo lineal. Los coristomas óseos epibulbares simples están compuestos de hueso en la superficie del globo ocular. Por lo general, se presentan en mujeres en el cuadrante superotemporal y no se asocian con otras afecciones.
> Granuloma piógeno
Un granuloma piógeno es una acumulación de tejido de granulación en el sitio de una lesión tisular previa. Es causado por inflamación, cirugía o traumatismo y aparece como una masa roja brillante elevada con sangre prominente. El granuloma de "oso de peluche" resulta de fibras sintéticas introducidas en el saco conjuntival, típicamente del fórnix inferior, y es particularmente común en niños debido al uso de mantas reconfortantes y juguetes de tela, que a menudo están cerca del ojo. Las opciones de tratamiento incluyen observación, corticosteroides tópicos o escisión quirúrgica.
> Papiloma
Un papiloma conjuntival es un tumor fibrovascular benigno con una apariencia rosada similar a una coliflor, que se presenta con mayor frecuencia en el fórnix inferior y se asocia con el virus del papiloma humano. Puede causar sensación de cuerpo extraño, cierre incompleto del párpado o desgarros hemorrágicos. Los papilomas grandes o sintomáticos se pueden extirpar o tratar con crioterapia. La cimetidina oral también puede ser eficaz.
> Nevos conjuntivales
Los nevos conjuntivales son los tumores melanocíticos más comunes de la superficie ocular. Tienen pigmentación variable, a menudo con cambios quísticos claros, y por lo general se presentan cerca del limbo. El crecimiento y el aumento de la pigmentación pueden ocurrir en la adolescencia y no necesariamente significan transformación maligna. Los nevos deben controlarse con comparación fotográfica; menos del 1% se malignizan.
> Melanocitosis oculodérmica congénita
La melanocitosis oculodérmica congénita es una melanocitosis benigna que sigue la distribución de la rama V1 o V2 del nervio trigémino, y produce una pigmentación gris azulada de la conjuntiva y la esclerótica. Cuando afecta la piel periocular, se denomina nevo de Ota. Por lo general, se presenta al nacer y en pacientes de ascendencia asiática y africana. Existe un mayor riesgo de glaucoma, así como un aumento significativo del riesgo de formación de melanoma uveal a 1 en 400, por lo que se requiere un control anual.
| TUMORES ORBITARIOS |
Los tumores orbitarios en niños son raros; el 95% de los biopsiados son benignos, siendo las lesiones quísticas y vasculares las más comunes.
> Presentación
Un tumor orbitario se presenta con hallazgos inespecíficos. Las lesiones superficiales que afectan la órbita anterior pueden visualizarse subcutáneamente como un nódulo palpable. Las lesiones orbitarias más profundas a menudo son más difíciles de diagnosticar. Los signos de lesiones que ocupan espacio en la órbita incluyen proptosis o protrusión del globo ocular, desplazamiento del globo ocular, estrabismo, ptosis palpebral e hinchazón y decoloración de los párpados. La disminución de la visión puede ocurrir con la compresión del nervio óptico por efecto de masa de las lesiones posteriores. Los signos orbitarios pueden aparecer de forma aguda en horas o días o lentamente con el tiempo, dependiendo de la etiología. Ocasionalmente, una neoplasia maligna puede imitar a la celulitis orbitaria y debe sospecharse cuando un paciente presenta proptosis y edema y ningún otro signo de inflamación. El hematoma periorbitario y la equimosis sin trauma también deben despertar la preocupación por una posible neoplasia maligna. Los hallazgos característicos de las enfermedades orbitarias se presentan en la Tabla 2.
> Examen
Además de la evaluación de la visión y pupilar, la evaluación de los tumores orbitarios debe incluir el control del déficit de movimiento extraocular, estrabismo y proptosis o desplazamiento del ojo. La proptosis se puede identificar fácilmente mediante una "visión a ojo de gusano", inclinando la cabeza del paciente hacia arriba y mirando desde abajo. La proptosis verdadera debe diferenciarse de la pseudoproptosis, que puede ser causada por caída o retracción del párpado, tamaño orbital asimétrico, deformidad congénita o agrandamiento del globo ocular o de la córnea. La dirección del desplazamiento del globo ocular y las características de los cambios periorbitales son útiles para limitar el diagnóstico diferencial (Tabla 3). La visión del color y los campos visuales de confrontación son pruebas aditivas para evaluar la función del nervio óptico. A menudo se necesitan imágenes de alta calidad con tomografía computarizada (TC) o resonancia magnética (RM) que pueden ayudar a diferenciar masas. La ecografía puede ser útil y puede realizarse rápidamente en un niño poco cooperativo sin sedación. El diagnóstico definitivo a menudo requiere biopsia.
> Quistes dermoides y epidermoides
Los quistes dermoides son la lesión orbitaria más común de la infancia. Se presentan como un crecimiento indoloro, lento, bien circunscrito y móvil en el borde orbital superolateral cerca de la sutura cigomático-frontal. El examen clínico suele ser suficiente para el diagnóstico. La ruptura del quiste puede provocar una respuesta inflamatoria robusta; las lesiones suelen extirparse para evitar esta complicación.
> Hemangioma infantil (hemangioma capilar)
El hemangioma infantil es el tumor vascular infantil más común de la órbita. Los hemangiomas infantiles suelen presentarse en las primeras semanas de vida con una fase inicial de crecimiento rápido seguida de estabilidad e involución en los años siguientes. Los tumores superficiales tienen un aspecto rojo brillante y abultado, y los hemangiomas más profundos aparecen azulados. El diagnóstico se basa en la apariencia clínica, pero en escenarios ambiguos, las imágenes son útiles. Las lesiones faciales grandes deben generar sospechas de síndrome PHACE, que incluye formaciones de la fosa posterior, hemangiomas faciales, lesiones arteriales, anomalías cardíacas y anomalías oculares. La observación es razonable si no hay efecto sobre la visión. El compromiso visual o la amenaza a la visión requieren tratamiento. El maleato de timolol tópico (solución formadora de gel al 0,5% 4-5 veces al día durante 6-9 meses) o la terapia con láser de colorante pulsado han demostrado eficacia para lesiones superficiales, y el propranolol oral (2-3 mg/kg al día) es el preferido para lesiones más grandes y profundas.(30)
> Malformaciones vasculares
Las malformaciones vasculares incluyen malformaciones venosas, malformaciones arteriovenosas y linfangiomas. La historia clínica, el examen físico y las imágenes son útiles para distinguir las lesiones y ayudar en el diagnóstico. Las malformaciones vasculares que se encuentran en la órbita anterior se presentan como masas blandas y azuladas. Las que son posteriores pueden presentarse como proptosis.
Las venas orbitarias que se dilatan a través de una debilidad en la pared vascular se denominan várices orbitarias. Estas lesiones de bajo flujo ocurren durante toda la infancia, con mayor frecuencia en la adolescencia. Los cambios en la presión venosa (es decir, Valsalva) pueden causar el llenado de la lesión y proptosis. Las complicaciones incluyen trombosis que produce dolor, proptosis y disminución de la visión; hemorragia orbitaria aguda; y disminución de la visión por compromiso del nervio óptico. Las imágenes ayudan al diagnóstico y la maniobra de Valsalva durante la toma de imágenes puede ser útil. Existe riesgo de malformaciones vasculares intracraneales no contiguas, por lo que se requiere evaluación cerebral.
Las malformaciones linfáticas generalmente se presentan en la primera década de vida. Contienen componentes venosos y linfáticos y se cree que son una variante de las várices orbitarias.(33) Las malformaciones linfáticas pueden agrandarse durante una enfermedad viral aguda o en el contexto de una hemorragia espontánea, lo que puede causar una proptosis dolorosa y aguda. La celulitis orbitaria recurrente precipitada por una enfermedad viral debe generar sospecha de linfangioma oculto. Las opciones de tratamiento son la observación, el tratamiento sistémico con sildenafil o sirolimus, la escisión completa o parcial y la escleroterapia.
> Rabdomiosarcoma
El rabdomiosarcoma es el sarcoma infantil más común y la neoplasia maligna orbitaria primaria pediátrica más frecuente. El 10% de los casos de rabdomiosarcoma se originan en la órbita. También puede presentarse como rabdomiosarcoma orbitario secundario. La distribución por edad es bimodal (alcanza un máximo a los 4 y 14 años), y el grupo de edad más joven tiene más probabilidades de presentar tumores orbitarios. La presentación clásica es la proptosis unilateral progresiva de aparición rápida. El desplazamiento del globo ocular, el edema palpebral y la ptosis se presentan en la mayoría de los pacientes. Pocos pacientes tendrán una masa palpable y dolor en la presentación.Es imperativo diferenciar un caso sospechoso de celulitis orbitaria agresiva del rabdomiosarcoma porque estos diagnósticos pueden compartir una superposición clínica extensa. El tratamiento puede implicar quimioterapia y radiación junto con cirugía. El pronóstico depende de la morfología; el embrionario es el subtipo orbitario más común con mejor pronóstico, mientras que el subtipo alveolar tiende a tener un peor pronóstico.
> Linfoma
Los linfomas orbitarios son poco frecuentes en pediatría. El linfoma de Burkitt es el tipo orbitario más común y es típicamente unilateral, pero puede presentarse en forma bilateral. Los niños pueden presentar síntomas generales junto con signos oculares. Para el diagnóstico se requieren estudios de imagen y un examen ocular completo seguidos de una biopsia. Los regímenes de tratamiento incluyen quimioterapia sistémica con o sin radioterapia adyuvante. El pronóstico es bueno si se trata de manera oportuna.
> Leucemia y sarcoma granulocítico
La leucemia mieloide aguda es el tipo más común de leucemia que afecta la órbita y es la etiología más común de proptosis bilateral en niños. La afectación de la órbita puede ser el primer signo de la enfermedad. La leucemia mieloide aguda orbitaria se presenta típicamente con edema periorbitario bilateral y proptosis, aunque se observan presentaciones unilaterales en el 40% de los niños. La proliferación de células granulocíticas extramedulares puede dar lugar a una masa discreta, denominada sarcoma granulocítico (mieloide), y es un presagio de leucemia mieloide aguda. Otros hallazgos extraoculares incluyen hemorragia subconjuntival, quemosis conjuntival, edema palpebral y restricción extraocular. Pueden estar presentes manifestaciones constitucionales, linfadenopatía, hepatoesplenomegalia y dolor óseo. La afectación orbitaria se ha asociado con un peor pronóstico, lo que hace que el diagnóstico rápido sea imperativo para obtener buenos resultados.
> Meningioma de la vaina del nervio óptico
Los meningiomas de la vaina del nervio óptico son poco frecuentes en la población pediátrica, pero pueden estar asociados con la neurofibromatosis tipo 2.(45)(46) Causan pérdida de la visión monocular indolora y gradualmente progresiva. La obtención de imágenes por resonancia magnética o TC es crucial para el diagnóstico; la TC muestra clásicamente una vaina del nervio óptico engrosada de forma difusa con áreas de engrosamiento paralelas al nervio óptico conocidas como configuración en “vía de tren”. Se cree que los meningiomas de la vaina del nervio óptico pediátricos se comportan de forma más agresiva que en los adultos, con un mayor potencial maligno. La extensión intracraneal es más común en niños que en adultos.(45) Por lo tanto, las lesiones suelen extirparse como primera modalidad de tratamiento en niños. En algunos casos, la enfermedad no es progresiva y se puede observar.
> Glioma del nervio óptico (glioma de la vía óptica, astrocitoma pilocítico juvenil)
Los gliomas del nervio óptico pediátricos son astrocitomas pilocíticos de bajo grado y son el tumor del sistema nervioso central más común en la infancia. A menudo se presentan en niños con neurofibromatosis tipo 1 (NF1), pero pueden ocurrir esporádicamente. La edad media de presentación es de 9 años. Los niños presentan pérdida de visión ipsilateral y proptosis que aumenta lentamente. La resonancia magnética muestra un agrandamiento bien circunscrito del nervio óptico. El crecimiento de la lesión es impredecible y el manejo es un desafío. La observación es apropiada; sin embargo, si la visión está amenazada, está indicado el manejo con quimioterapia o terapia molecular dirigida. En casos raros de pérdida rápida de la visión o proptosis progresiva, puede ser necesaria una desbulminación quirúrgica.
> Neurofibromas
El neurofibroma puede aparecer en el párpado como un neurofibroma plexiforme, lo que sugiere neurofibromatosis. Los neurofibromas plexiformes se describen como de consistencia de “bolsa de gusanos” y producen una curva en forma de S en el párpado superior. En raras ocasiones, los neurofibromas pueden sufrir una transformación maligna. El tratamiento es la observación o, en casos de pérdida de visión o deformidad cosmética, la reducción quirúrgica.
> Enfermedad metastásica en la órbita
La órbita es el sitio más común de metástasis oculares en niños, mientras que en adultos la úvea es la zona principal afectada. El neuroblastoma, una neoplasia maligna infantil de origen en la cresta neural, es el tumor sólido extracraneal más común en niños.(50) Hace metástasis en la órbita a través de propagación hematógena en el 15% de los casos y es el tipo más común de tumor orbitario metastásico en pacientes pediátricos. La presentación orbitaria comúnmente implica equimosis y proptosis palpebral bilateral. Comúnmente denominados “ojos de mapache”, los hallazgos son el resultado de la obstrucción de los vasos sanguíneos palpebrales por metástasis orbitarias. La mayoría de los pacientes tienen un tumor suprarrenal primario en el momento de la presentación orbitaria; sin embargo, el 3% presenta metástasis orbitarias como primer signo de enfermedad. Otros hallazgos oculares del neuroblastoma pueden incluir el síndrome de Horner resultante de la interrupción de la vía simpática y el opsoclono, un síndrome paraneoplásico que se presenta con ojos “bailarines”.
El sarcoma de Ewing y el tumor de Wilms se metastizan con menos frecuencia en la órbita en los niños.
> Lesiones que simulan tumores orbitarios
Enfermedad ocular tiroidea. La enfermedad ocular tiroidea (EOT) es rara en la población pediátrica. Es más común en mujeres y se presenta a una edad media de 12 años. La mayoría de los pacientes desarrollan EOT en un estado hipertiroideo al mismo tiempo que la aparición de la enfermedad. La presentación más común de la EOT pediátrica es la proptosis seguida de retracción palpebral. Los pacientes pediátricos con EOT tienen síntomas más leves que los adultos. El manejo estricto de los niveles de hormonas tiroideas es crucial, pero la enfermedad no siempre se correlaciona con los niveles hormonales. En casos raros, se requiere el uso de corticosteroides o descompresión quirúrgica de la órbita ante pérdida de visión inminente. El tocilizumab, un antagonista del receptor de interleucina-6, se ha utilizado con éxito en un caso de EOT pediátrica progresiva.
Celulitis orbitaria. La celulitis orbitaria es una infección que puede poner en peligro la visión y la vida, situada detrás del tabique orbitario. Por lo general, es secundaria a sinusitis, pero puede ocurrir en el contexto de una enfermedad odontogénica, una infección hematógena o una infección palpebral o facial. En casos avanzados, se pueden observar abscesos subperiósticos. Puede ser necesario realizar estudios de imagen orbitarios especializados, especialmente si se observa pérdida visual, motilidad extraocular reducida, proptosis, pérdida de la visión del color, defecto pupilar aferente, enfermedad bilateral o falta de respuesta a los antibióticos. El inicio rápido de antibióticos intravenosos de amplio espectro es crucial, y los corticosteroides intravenosos pueden ser beneficiosos en la población pediátrica. También puede ser necesario el control de la fuente con cirugía de los senos nasales. Las neoplasias malignas, como el rabdomiosarcoma y el retinoblastoma, pueden enmascararse como celulitis orbitaria.
Inflamación orbitaria idiopática (pseudotumor orbitario, inflamación orbitaria no específica). La inflamación orbitaria idiopática describe una lesión orbitaria que ocupa espacio sin etiología infecciosa ni características histopatológicas compatibles con un trastorno inflamatorio conocido. Aunque es común en adultos, es rara en pediatría. La inflamación orbitaria idiopática se presenta típicamente con signos orbitarios; los niños tienen más probabilidades de tener enfermedad bilateral y síntomas constitucionales concomitantes. Un diagnóstico de exclusión, una resonancia magnética y una evaluación serológica son útiles para eliminar diagnósticos alternativos. La mayoría de los casos se tratan con éxito con corticosteroides en dosis altas, seguidos de una reducción prolongada, aunque se informa recurrencia en hasta el 75% de los casos pediátricos. Los agentes inmunomoduladores se consideran tempranamente para evitar el uso prolongado de corticosteroides en niños.
| TUMORES INTRAOCULARES |
> Presentación
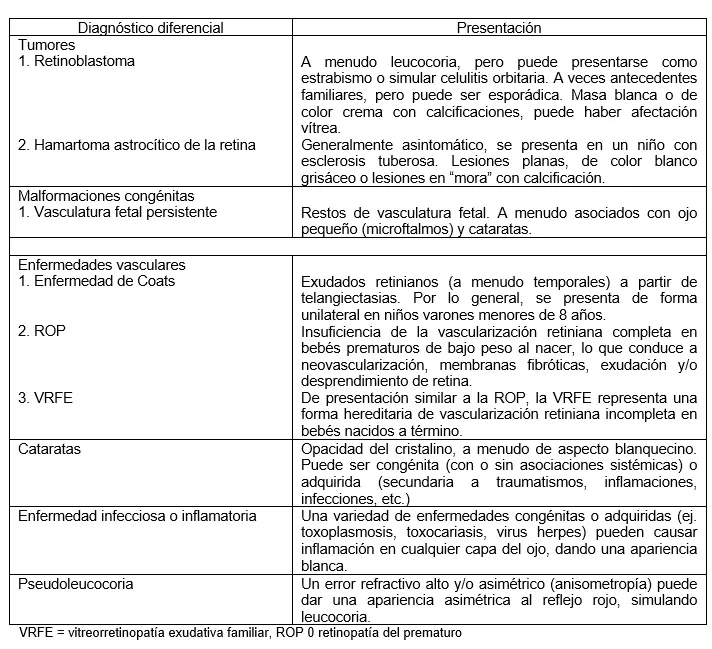
La presentación de los tumores intraoculares depende del tamaño del tumor y de su ubicación en el ojo. Los tumores pequeños o periféricos pueden ser asintomáticos y encontrarse solo en un examen de rutina del fondo de ojo. Los tumores más grandes o los que afectan la visión pueden causar leucocoria, estrabismo y disminución de la visión; el diagnóstico diferencial de la leucocoria se presenta en la Tabla 4.
Los pacientes con un tumor intraocular y mala visión desde la infancia temprana pueden presentar nistagmo.
> Examen
Los tumores intraoculares del segmento anterior, como los que afectan el iris, pueden visualizarse en la inspección, pero a menudo requieren aumento con una lámpara de hendidura. Los tumores posteriores del cuerpo ciliar, la retina y la coroides requieren un examen del fondo de ojo. Las pruebas oftálmicas con ecografía, tomografía de coherencia óptica y angiografía con fluoresceína pueden ser útiles.
> Hamartoma del iris (nódulo de Lisch)
Los nódulos de Lisch son hamartomas melanocíticos de 1 a 2 mm, de color tostado o marrón, de la superficie del iris. Por lo general, son asintomáticos y no causan trastornos visuales ni requieren tratamiento. Son marcadores patognomónicos de la NF1. Las lesiones rara vez se presentan durante la infancia, pero aparecen durante los años de la niñez, y el 100% de los pacientes con NF tienen nódulos de Lisch en la edad adulta temprana.(62) Por lo general, aparecen antes que los neurofibromas y, por lo tanto, pueden ser útiles para confirmar el diagnóstico. Otros hallazgos oftálmicos característicos de la NF1 incluyen gliomas del nervio óptico y displasia del ala esfenoidal.
> Meduloepitelioma
Los meduloepiteliomas intraoculares son tumores congénitos poco frecuentes del epitelio del cuerpo ciliar.Se presentan como una masa carnosa o quística en el cuerpo ciliar y pueden conducir a complicaciones como subluxación del cristalino, formación de cataratas o glaucoma neovascular. Generalmente se hacen evidentes entre la primera y la segunda década de vida, y se presume que los adultos que son diagnosticados recientemente tienen tumores de crecimiento lento o transformación maligna de un tumor congénito. El 50% de los meduloepiteliomas son malignos, pero la extensión extraocular y la metástasis son raras. Rara vez pueden asociarse con el síndrome de predisposición al cáncer hereditario DICER-1. El tratamiento incluye resección local, braquiterapia de placa y enucleación.
> Hamartoma astrocítico
Los hamartomas astrocíticos son proliferaciones benignas de células gliales que surgen de la capa de fibras nerviosas de la retina.Pueden surgir esporádicamente pero se asocian con esclerosis tuberosa y son un criterio para establecer el diagnóstico. También se han descripto hamartomas astrocíticos en la NF1. La mayoría de los casos se presentan en la adolescencia o en la adultez temprana.
Los tumores pueden tener varias formas diferentes, desde masas planas, sésiles y translúcidas hasta tumores nodulares tipo “morera” con calcificaciones. Los pacientes con esclerosis tuberosa y hamartomas astrocíticos tienen mayor probabilidad de tener tumores intracraneales y extracraneales, como hamartomas subependimarios de células gigantes y angiomiolipomas renales. Los hamartomas astrocíticos suelen ser asintomáticos, pero tienen el potencial de ejercer tracción sobre la retina y causar una acumulación visualmente significativa de líquido subretinal según el tamaño y la proximidad a la mácula. En casos que amenazan la visión, se ha demostrado que los inhibidores de mTOR sirolimus y everolimus reducen la exudación y el tamaño de la lesión.
Nevos uveales. Los nevos uveales son proliferaciones benignas de melanocitos del iris y la coroides posterior.Son asintomáticos y su tamaño se mantiene estable con el tiempo.
Hipertrofia congénita del epitelio pigmentario de la retina. La hipertrofia congénita del epitelio pigmentario de la retina (HCEPR) son lesiones benignas, planas y de pigmentación oscura de la retina que están presentes desde el nacimiento. Tienen lagunas no pigmentadas características con un halo circundante y se diagnostican de manera incidental en el examen del fondo de ojo. La presencia de HCEPR pisciformes pequeñas, bilaterales o múltiples se asocia con poliposis adenomatosa familiar y los pacientes tienen un alto riesgo de cáncer de colon.
Melanocitoma. Los melanocitomas son lesiones benignas que se encuentran típicamente en el disco óptico, donde aparecen profundamente pigmentados y se diagnostican fácilmente en el examen del fondo de ojo. Aunque la mayoría se diagnostican en la edad adulta, algunos niños desde pequeños presentan estas lesiones benignas. El crecimiento y las complicaciones, incluyendo glaucoma y pérdida de la visión, pueden ocurrir rara vez. La transformación maligna ocurre en el 1% al 2% de las lesiones.
Melanoma. El melanoma uveal es raro en niños y compone menos del 1% de todos los melanomas uveales. La melanocitosis ocular, descripta previamente en este documento, aumenta el riesgo de melanoma uveal, y los pacientes con esta afección deben ser controlados regularmente durante toda la vida.
Retinoblastoma. El retinoblastoma es la neoplasia maligna intraocular más común en la infancia. Es causado por una mutación de pérdida de función en el gen RB1 en el cromosoma 13. Las mutaciones esporádicas conducen a una enfermedad unilateral, y las mutaciones hereditarias de la línea germinal suelen causar enfermedad bilateral. Los pacientes con mutaciones de la línea germinal tienen riesgo de otros tumores, incluidos pineoblastomas y sarcomas secundarios. El retinoblastoma suele presentarse en niños antes de los 4 años de edad. Aproximadamente el 30% de los casos se deben a mutaciones de la línea germinal. Es fundamental que estos pacientes y sus familias reciban asesoramiento genético adecuado. Todos los familiares de primer grado de un paciente con retinoblastoma de la línea germinal deben ser examinados lo antes posible después del diagnóstico y luego recibir exámenes de fondo de ojo con dilatación cada 1 a 2 años, de manera indefinida.
El signo de presentación más común es la leucocoria, seguida de estrabismo e inflamación orbitaria o pseudocelulitis. El estándar de atención para los tumores avanzados es la enucleación. Los tumores más pequeños se pueden tratar con métodos de salvamento del globo, que incluyen quimioterapia intravítrea, quimioterapia intravenosa y quimioterapia intraarterial, una técnica más nueva que administra quimioterapia a través de un microcatéter directamente en la arteria oftálmica. También se pueden utilizar otros tipos de tratamiento focal, como láser, crioterapia y radioterapia en placa. En los países desarrollados, la muerte por retinoblastoma es rara.
Hemangioma coroideo. Los hemangiomas coroideos son tumores vasculares benignos, congénitos, que se presentan en 2 formas, circunscrita y difusa, la última asociada con el síndrome de Sturge-Weber. Cuando son asintomáticos, se pueden controlar. Si ocurren complicaciones visuales, como desprendimiento de retina, las opciones de tratamiento incluyen láser, radiación y/o inyecciones anti factor de crecimiento endotelial vascular.
Hemangioblastoma retiniano. El hemangioblastoma retiniano es un tumor vascular benigno poco frecuente con una arteria nutricia dilatada característica y una vena de drenaje tortuosa. El hemangioblastoma retiniano está altamente asociado con la enfermedad de von Hippel-Lindau, una enfermedad autosómica que produce una variedad de tumores benignos y malignos. Los pacientes con hemangioblastoma retiniano deben someterse a una evaluación para la enfermedad de von Hippel-Lindau.(96) La afectación macular, la fuga de exudado y líquido subretinal, y el sangrado pueden provocar pérdida de la visión; las opciones de tratamiento incluyen terapia láser y radiación. Los primeros estudios han demostrado que el belzutifán, un inhibidor de HIF-2a recientemente aprobado para el tratamiento de tumores asociados a von Hippel-Lindau en pacientes mayores de 18 años, conduce a la regresión de los hemangioblastomas retinianos.
| Comentario |
Los tumores oftálmicos en pediatría se presentan al nacer o durante la infancia y presentan etiología variada. La mayoría son benignos, pero los malignos pueden ser potencialmente mortales. Ambos tipos de tumores pueden causar pérdida de la visión por afectación de estructuras oculares y factores ambliogénicos. Ciertos tumores, en particular los vasculares y neurogénicos, pueden asociarse con enfermedad sistémica, requiriendo evaluaciones adicionales. El pediatra debe estar familiarizado con los diagnósticos y presentaciones comunes de los tumores oftálmicos, ya que el diagnóstico y tratamiento precoces pueden optimizar los resultados generales a corto y largo plazo.
Tabla 1. Afecciones no oftálmicas asociadas con tumores malignos de párpados pediátricos

Tabla 2. Hallazgos característicos de las enfermedades oculares orbitarias
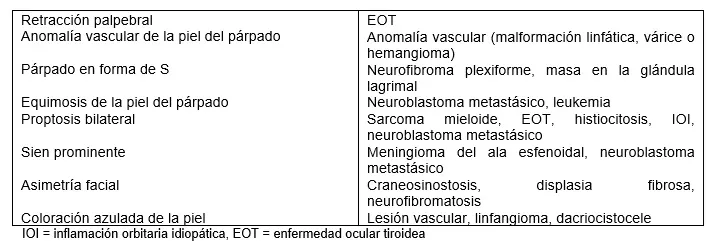
Tabla 3. Diagnóstico diferencial de proptosis
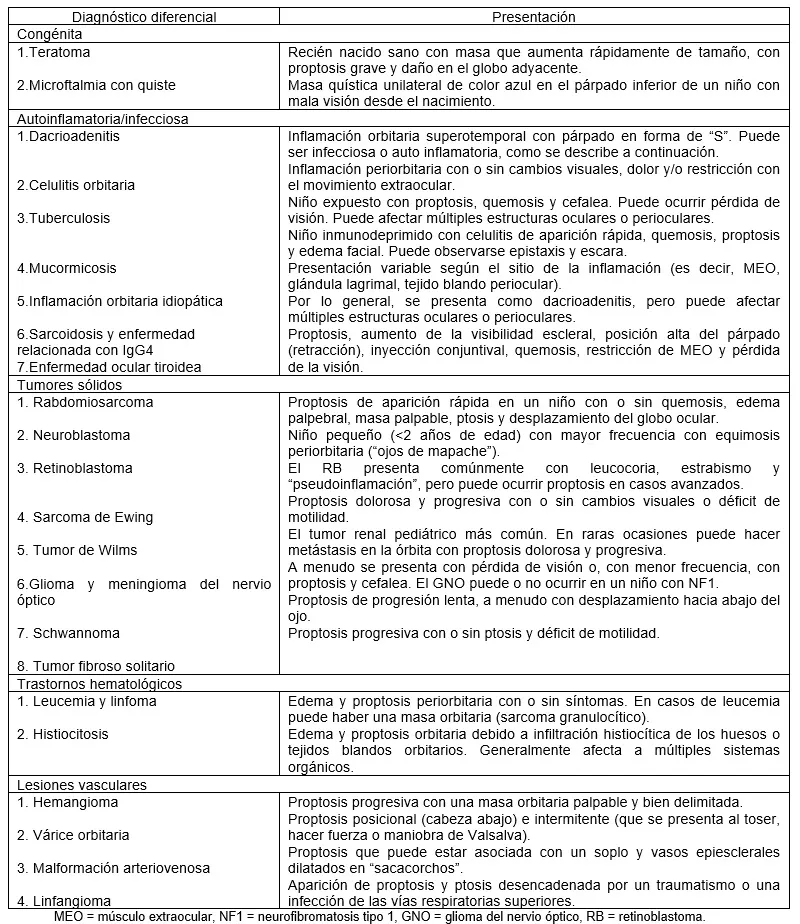
Tabla 4. Diagnóstico diferencial de leucocoria