Wells y Smith describieron en 1979 a la celulitis eosinofílica (síndrome de Wells, WS), y notaron que algunos pero no todos los pacientes con WS presentan eosinofilia. Los pacientes con eosinofilia idiopática persistente pueden rotularse dentro del síndrome hipereosinofílico (HES), que representa un espectro de desórdenes de severidad, causas y resultados variables.
En este artículo los autores proponen que los pacientes que presentan el espectro HES con hallazgos cutáneos de WS y sin enfermedad extracutáneas pueden clasificarse como hipereosinofilia persistente con síndrome de Wells (PHEWS).
Se presenta el caso de un paciente con WS que tiene HES sin manifestaciones extracutáneas y se propone un nuevo término y criterios diagnósticos para ésta condición.
Reporte del caso:
Se presenta una mujer de 64 años con antecedentes de erupción pruriginosa de 6 años de evolución. Las lesiones individuales persistían por varias semanas, dejando hiperpigmentación postinflamatoria, pero aparecían lesiones nuevas.
Al exámen físico, se observaban pápulas rojas, nódulos y lesiones urticarianas en tronco y miembros (Fig 1 a).
Al exámen histológico, la epidermis era normal. Se observan infiltrados perivasculares e intersticiales en dermis superficial, conteniendo numerosos eosinófilos y figuras en llamas ocasionales (Fig 1 b).
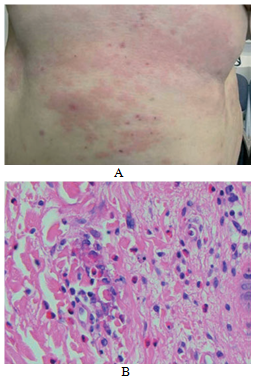
Figura 1 (a) Múltiples placas urticarianas en abdomen con áreas escoriadas;
(b) eosinófilos y macrófagos alrededor de un foco central de colágeno, recubierto en un material granular eosinofílico (H&E).
Las investigaciones de laboratorio identificaron hipereosinofilia presente durante la enfermedad, 4.12 x 109 eosinófilos por litro (rango normal 0-0.4 x 109 L) y continuaba en forma persistente 1.5 x 109L por 5 años. El hemograma completo, tests de función renal y hepática, perfil autoinmune, eritrosedimentación, proteina C reactiva, niveles séricos de IgE y triptasa eran normales.
Los tests serológicos para filariasis, esquistosomiasis y estrongiloides fueron negativas. No presentaba anormalidades en la radiografía de tórax. Los estudios citogenéticos y moleculares no idenificaron anormalidad hematológica clonal.
Tres años previos, al administrar un curso corto de corticoides se logró casi la remisión clínica completa con mejoría marcada del recuento de eosinófilos, con recaída posterior al suspender los corticoides.
En este caso, se realizó el diagnóstico clínico patológico de WS, sin embargo el paciente se encuadró en el espectro HES.
Wells y Smith describieron los hallazgos histopatológicos característicos de WS. Clásicamente y en su forma más severa, el WS se presenta inicialmente con pseudocelulitis aguda, que se puede extender para afectar una gran parte de un miembro. Las áreas son rojas y edematosas, puede haber ampollas, la piel afectada no tiene temperatura elevada a la palpación, y no responde a antibióticos. Las lesiones se hacen infiltradas, firmes y granulomatosas, con una discoloración verde-azulada, resolviendo en varias semanas, simulando a lesiones de morfea.
Wells y Smith luego describieron otras presentaciones cutáneas menos severas de WS, llamadas urticaria símil, ampollar, papular, nodular, anular y en placas, que generalmente son pruriginosas.
En los casos que presentaron, la eosinofilia en sangre periférica no siempre estaba presente, y no hubo comentarios de cuanto tiempo persistía la eosinofilia ( si estaba presente), o si recurría en el futuro.
Los hallazgos clínicos son múltiples en el WS, por lo que algunas veces el diagnóstico sólo puede hacerse por las características cutáneas e histológicas. Dichas características incluyen edema dérmico con infiltración de eosinófilos y macrófagos entre los haces de tejido conectivo, con figuras en llama (eosinófilos agrupados y macrófagos alrededor de un foco central de colágeno cubierto de un material granular eosinofílico).
Originariamente Wells y Smith describieron al WS como una probable reacción de hipersensibilidad inducida por varios desencadenantes como picaduras de insecto o reacciones a drogas. Desde su estudio, el WS se ha reportado que ocurre en enfermedades sistémicas asociadas con eosinofilia como Síndrome de Churg Strauss, HES y leucemia linfocítica crónica. Parece que el WS representa un patrón de reacción cutánea que se desarrolla en cualquier situación en la que el número anormal de eosinófilos se acumulan en la piel, por alguna razón, algunas veces desconocida.
El Síndrome hipereosinofílico es un término que involucra a un amplio rango de condiciones heterogéneas. Desde 1975, se han utilizado tres criterios para definir al HES: 1- eosinofilia en sangre mayor de 1.5 x 109 L por más de 6 meses o muerte en ese tiempo, asociadas a síntomas y signos de enfermedad hipereosinofílica; 2-sin causa secundaria de eosinofilia; 3-signos y síntomas de daño de órgano. En HES, la infiltración eosinofílica de los tejidos y liberación de mediadores ocasiona daño de órganos variable, que a pesar de un tratamiento agresivo, puede resultar en complicaciones severas y aún la muerte. Dichas complicaciones incluyen falla cardíaca y fibrosis, neuropatía periférica y disfunción generalizada del sistema nervioso central como signos de la neurona motora superior y encefalopatía. Manifestaciones pulmonares (fibrosis y tos no productiva), hiperesplenismo, hepatitis, obstrucción de la vena hepática, esofagitis eosinofílica, gastritis o colitis, y anormalidades coroidales.
Sin embargo, algunos pacientes con HES son asintomáticos o se presentan con una enfermedad más benigna, tal vez limitada a la piel, y estos pacientes pueden nunca desarrollar complicaciones, a pesar de la hipereosinofilia crónica.
Desde que fue descripto por primera vez, se han identificado las causas subyacentes en algunos casos de HES.
Para propósitos diagnósticos, no se insiste en que la hipereosinofilia sea verdaderamente idiopática, se tienen que excluir causas secundarias de eosinofilia como la inducida por drogas, por parásitos, causas alérgicas y autoinmunes.
Por ejemplo, los pacientes con HES mieloproliferativa (M)-HES pueden identificarse por técnicas moleculares y genéticas. La mutación cromosómica más común observada en M-HES es la delección en el cromosoma 4q12, causando fusión en los genes FIP1L1/ PDGFRA F/P, dando origen a una proteína con actividad tirosinaquinasa. Además, usando citometría de flujo se ha identificado HES-L (linfocítica), caracterizada por una población aberrante de células T de origen desconocido, que secretan citoquinas eosinofilopoyéticas, interleuquinas (IL)-3 e (IL)-5 causando eosinofilia.
En el 2005, se han propuesto 6 subtipos de HES: 1-M-HES (incluyendo F/P negativo con características mieloproliferativas , 2-L-HES (con población demonstrable de linfocitos fenotípicamente aberrantes o clonales), 3-eosinofilia familiar (incluyendo una forma autosómica dominante que mapea en el cromosoma 5q31.33, 4-HES no identificada (idiopático con o sin síntomas, incluyendo variantes episódicas, 5-superposición de HES (enfermedad eosinofílica restringida a un órgano o sistema con eosinofilia mayor de 1.5 x 109L, y 6- asociada a HES (eosinofilia mayor de 1.5 x 109L bajo otro diagnóstico, en el que la eosinofilia se ha descripto como un hallazgo en ese subtipo afectado).
Es importante identificar diferentes subtipos de HES para permitir un tratamiento con buenos resultados. Por ejemplo, los corticoides fueron inicialmente el tratamiento de primera línea para todos los tipos de HES, pero luego de identificar la mutación F/P, estos pacientes se tratan exitosamente con mesilato de imatinib.
El compromiso cutáneo está presente en el 45-60% de los pacientes con HES, y se ha descripto prurito, pápulas eritematosas, nódulos, lesiones símil urticaria y angioedema. El diagnóstico de WS que ocurre en dichos pacientes con HES puede ocurrir, e incluye el caso descripto. Wells y Smith reconocieron ésta superposición en 1979, y reconocieron a este cuadro como el “final benigno” del espectro HES (“benigno” indica sin complicaciones extracutáneas ocasionadas por la eosinofilia). Sin embargo, el tiempo ha demostrado que el término “benigno” es engañoso, ya que algunos pacientes con eosinofilia crónica pueden desarrollar enfermedad de órganos internos, incluyendo transformación a leucemia o linfoma.
Utilizando la clasificación actual, los pacientes con WS y eosinofilia idiopática persistente sin compromiso de órgano extracutáneos no se incluyen en ningún subtipo de HES.
Además, la eosinofilia, aunque generalmente está presente, no es necesaria para realizar el diagnóstico de WS, y por lo tanto los pacientes no deben diagnosticarse con WS solamente, especialmente si la eosinofilia es persistente. Es importante identificar y clasificar a dichos pacientes clínica e histopatológicamente, y debe definirse la etiología, tratamientos y resultados en dichos casos.
Los autores expusieron el caso para distinguir pacientes con WS de los que presentan WS más eosinofilia idiopática persistente sin compromiso de órganos extracutáneos. Estos casos son un subgrupo identificable de pacientes en el espectro HES. Actualmente no existe un sistema de clasificación que identifique adecuadamente a dichos pacientes, y por lo tanto los autores proponen una nueva clasificación de hipereosinofilia persistente con síndrome de Wells “PHEWS”, usando los siguientes criterios diagnósticos: 1-hipereosinofilia idiopática persistente mayor de 1.5 x 109L, 2-características clínico patológicas de WS, 3-la eosinofilia no produce compromiso de órganos extracutáneos.
“Idiopático” significa que se han excluído causas de M-HES y de eosinofilia secundaria. Esto incluye evaluación por hematología, incluyendo medición de triptasa sérica, análisis de FIP1L1/PDGFRA, y posible biopsia de médula ósea con estudio citogenético, molecular e histoquímico. Se utilizó el punto de corte de más de 1.5 x 109L por 6 meses, como lo establecieron los criterios diagnósticos de HES realizado por Chusid y col en 1975 para definir “hipereosinofilia persistente”.
Cuando se realizan las investigaciones de complicaciones extracutáneas de hipereosinofilia persistente los médicos deben guiarse en los hallazgos clínicos y síntomas con un bajo umbral de sospecha de compromiso cardiaco y pulmonar.
El tratamiento en casos confirmados de PHEWS debe tener como objetivo la mejora sintomática, y como en el WS, inicialmente se utilizan corticoides sistémicos con ajuste de dosis según la respuesta. Los corticoides tópicos sólos pueden ser suficientes.
Se han reportado efectivos a los antihistamínicos con, dapsona, ciclosporina, azatioprina, griseofulvina, tetraciclinas e interferon alfa como agentes ahorradores de corticoides en pacientes que necesitan tratamiento a largo plazo.
Es recomendable el seguimiento a largo plazo, ya que no es posible predecir si se va a producir algo daño por la hiperesosinofilia persistente o si se desarrollará una malignidad hematológica en el futuro. Se espera que al establecer a PHEWS como un síndrome diferente, se respondan esos interrogantes en el futuro.
¿Qué aporta éste artículo a la práctica dermatológica?:
El síndrome de Wells es un proceso reactivo en el que algunos de los pacientes presentan eosinofilia y puede localizarse dentro del espectro HES. Cualquier eosinofilia en el contexto de WS debe investigarse, especialmente si es persistente.
Los pacientes pueden diagnosticarse de PHEWS si tienen hipereosinofilia idiopática, persistente más las características clínico patológicas de WS sin evidencia de compromiso de órganos extracutáneos mediados por eosinófilos. Es importante identificar a éstos pacientes a largo plazo y en el futuro deben definirse estrategias de manejo y tratamiento.
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodriguez Rivello