|
Varón de 21 años Enfermedad actual: Comienza 2 meses previos al ingreso con mialgias en ambos miembros inferiores y tronco, de intensidad máxima, que impiden la marcha, asociado a registros febriles diarios de hasta 40°C, tos productiva sin expectoración, dolor abdominal en hipogastrio, intermitente, de tipo cólico. Al interrogatorio dirigido refiere episodios de hematuria, gingivorragia, proctorragia y petequias, que se autolimitan. Niega sudoración nocturna por fuera de episodios febriles, pérdida de peso, prurito, contacto con roedores y síntomas urinarios. Por dicho cuadro cursó internación por 19 días en hospital de su ciudad de origen, donde continuó con registro febriles diarios asociado desde su inicio a pancitopenia, falla renal y alteraciones en el hepatograma. En dicha institución realizó 8 días de tratamiento antibiótico empírico con ampicilina-sulbactam y posteriormente 9 días de ceftriaxona y vancomicina. Por persistencia del cuadro clínico es derivado a nuestro nosocomio para continuar estudio y tratamiento. |
| Antecedentes personales |
- Síndrome de Asperger, sin tratamiento
- Asma desde la infancia. En tratamiento con salbutamol según necesidad
- Tabaquista ocasional.
- Niega otros hábitos tóxicos.
- Niega alergias.
| Examen físico |
> Paciente vigil, orientado globalmente. Impresiona moderadamente enfermo.
> Palidez cutáneo-mucosa generalizada.
> Signos vitales: PA: 100/60 mmHg, FC: 104 lpm, FR: 18 rpm, T°36.2 °C, saturación 97% (0.21).
> Cabeza y cuello: normocéfalo. Conjuntivas pálidas, escleras blancas. Movimientos oculares completos, no dolorosos. Pupilas isocóricas reactivas y simétricas. Fosas nasales permeables sin secreciones ni epistaxis. Puntos sinusales negativos. Mucosas semihúmedas, piezas dentarias incompletas en regular estado. Lengua central y móvil. Cuello cilíndrico, simétrico, se palpan adenopatías submaxilaes, móviles, indoloras, duroelásticas de 1 cm x 1 cm aproximadamente, a predominio derecho. Ingurgitación yugular 2/6 con colapso completo.
> Tórax: diámetro anteroposterior conservado, sin cicatrices. Columna sonora sin desviaciones, indolora. Puño percusión negativa bilateral.
> Aparato cardiovascular: no se observan latidos patológicos. No se palpa frémito. R1-R2 normofonéticos. Ritmo regular. No se auscultan soplos. Sin R3-R4.
> Aparato respiratorio: respiración costoabdominal, sin reclutamiento ni tiraje. Buena entrada bilateral de aire sin ruidos agregados. Excursión de bases conservada. Expansión de bases y vértices conservada.
> Abdomen: plano, blando, depresible, doloroso a la palpación profunda en fosa ilíaca izquierda e hipocondrio derecho sin defensa ni descompresión. No se palpan visceromegalias. Ruidos hidroaéreos positivos. Traube libre. Timpanismo conservado. No ausculto soplos.
> Miembros: edemas en los 4 miembros, godet 1 +. Tono, trofismo y sensibilidad conservados. Fuerza disminuida por dolor. Pulsos conservados. Diferencia de diámetro de 2 cm a predominio del miembro inferior derecho, sin eritema ni aumento de temperatura local. Sin adenopatías. Homans y Ollow negativos.
> Neurológico: funciones superiores conservadas. Pares craneales, motilidad activa y sensibilidad conservada. Equilibro y marcha conservados. Signos meníngeos negativos.
> Genitales: no presenta lesiones ni secreción en pene, testículos en bolsa.
| Exámenes complementarios previos |
|
LABORATORIO |
Día 1 |
Día 9 |
Día 10 |
Día 12 |
Día 18 |
|
Hemoglobina (g/dl) |
11.6 |
10.1 |
9.2 |
10.6 |
8.1 |
|
Hematocrito (%) |
32.8 |
29 |
26.9 |
30.1 |
23.5 |
|
Leucocitos (cel/mm3) |
2000 |
2900 |
2800 |
3500 |
2200 |
|
Neutrófilos |
73% |
77% |
74% |
72% |
74% |
|
Linfocitos |
20% |
17% |
22% |
19% |
15% |
|
Eosinófilos |
2% |
2% |
1% |
3% |
3% |
|
Basófilos |
0% |
0% |
0% |
0% |
0% |
|
Monocitos |
5% |
4% |
3% |
6% |
8% |
|
Plaquetas (cél/mm3) |
104000 |
161000 |
130000 |
140000 |
79000 |
|
Glicemia (mg/dl) |
87 |
114 |
- |
126 |
105 |
|
Uremia (mg/dl) |
96 |
33 |
38 |
38 |
36 |
|
Creatininemia (mg/dl) |
2.4 |
1.47 |
1.4 |
1.5 |
1.8 |
|
Bilirrubina total (mg/dl) |
0.9 |
1.6 |
1.51 |
- |
0.9 |
|
Bilirrubina indirecta (mg/dl) |
0.6 |
1.5 |
1.44 |
- |
0.61 |
|
Bilirrubina directa (mg/dl) |
0.3 |
0.05 |
0.07 |
- |
0.38 |
|
GOT (UI/l) |
448 |
298 |
- |
- |
188 |
|
GPT (UI/l) |
296 |
130 |
- |
- |
262 |
|
FAL (UI/L) |
- |
648 |
- |
- |
- |
|
Natremia (mEq/l) |
130 |
136 |
137 |
138 |
- |
|
Potasemia (mEq/l) |
4.42 |
4.24 |
4.62 |
4.93 |
- |
|
Cloremia (mEq/l) |
110 |
- |
- |
- |
- |
|
TP (segundos) |
12.6 |
- |
- |
- |
- |
|
KPTT (segundos) |
42 |
- |
- |
- |
- |
|
CPK (UI/l) |
1727 |
4585 |
6640 |
7980 |
1290 |
|
LDH (UI/l) |
- |
- |
1731 |
246 |
1771 |
|
Proteínas totales (g/l) |
- |
- |
4.10 |
- |
- |
|
Albúmina (g/l) |
- |
- |
4.9 |
- |
- |
- Frotis de sangre periférica: glóbulos blancos: 2800. 75% polimorfonucleares. Granulaciones tóxicas. No se observan blastos. Plaquetas 92000/mm3. Target cells.
- Serología Leptospirosis: anticuerpos (TR) positivo débil.
- Serología para HIV: negativo.
- Serologías para CMV y VEB: no reactivo.
- Serología para Fiebre Hemorrágica Argentina: negativo.
- Látex para artritis reumatoidea: negativo.
- FAN: positivo (1/640). Imagen homogénea.
- TSH: 6 uUI/ml.
- Cuantificación C3: 16 mg/dl.
- Cuantificación C4: 2 mg/dl.
- Ecocardiograma: sin hallazgos patológicos.
- Ecografía abdominal: esplenomegalia 170 mm. Escaso líquido libre en fondo de saco de Douglas.
Exámenes complementarios durante la internación:
|
LABORATORIO |
Día 0 |
Día 1 |
Día 2 |
Día 3 |
Día 4 |
Día 5 |
|
Hemoglobina (g/dl) |
8.6 |
7 |
6.4 |
6.1 |
7.3 |
6.8 |
|
Hematocrito (%) |
25.3 |
21.3 |
19.6 |
18.9 |
22.8 |
20.8 |
|
Leucocitos (cel/mm3) |
2800 |
1250 |
1400 |
1840 |
2220 |
3630 |
|
Neutrófilos % |
75 |
62 |
67 |
66 |
- |
- |
|
Linfocitos % |
20 |
24 |
20 |
20 |
- |
- |
|
Basófilos% |
- |
0 |
0 |
1 |
- |
- |
|
Eosinófilos% |
- |
4 |
4 |
7 |
- |
- |
|
Monocitos % |
- |
6 |
7 |
5 |
- |
- |
|
Plaquetas (cél/mm3) |
31000 |
73000 |
82000 |
89000 |
93000 |
92000 |
|
Glicemia (mg/dl) |
90 |
71 |
82 |
75 |
77 |
96 |
|
Uremia (mg/dl) |
53 |
65 |
71 |
74 |
72 |
86 |
|
Creatininemia (mg/dl) |
2.14 |
2.49 |
2.87 |
3.13 |
3.51 |
4.08 |
|
Bilirrubina total (mg/dl) |
0.85 |
0.69 |
0.76 |
0.70 |
0.91 |
1.34 |
|
Bilirrubina indirecta (mg/dl) |
0.07 |
0.02 |
- |
0.11 |
- |
0.22 |
|
Bilirrubina directa (mg/dl) |
0.78 |
0.67 |
- |
0.59 |
- |
1.22 |
|
GOT (UI/l) |
261 |
301 |
2.93 |
237 |
190 |
210 |
|
GPT (UI/l) |
81 |
80 |
78 |
70 |
64 |
73 |
|
Fosfatasa alcalina (UI/l) |
240 |
248 |
235 |
221 |
224 |
201 |
|
Gama glutamil transpeptidasa (UI/l) |
389 |
451 |
475 |
437 |
436 |
376 |
|
Pseudocolinesterasa (UI/l) |
2500 |
2462 |
2398 |
2694 |
3090 |
2504 |
|
Amilasa (UI/l) |
644 |
754 |
834 |
821 |
880 |
881 |
|
Proteínas totales (g/dl) |
4.8 |
- |
- |
4.4 |
- |
4.8 |
|
Albúmina (g/dl) |
2.6 |
- |
- |
2.5 |
- |
2.5 |
|
Natremia (mEq/l) |
139 |
142 |
139 |
138 |
137 |
136 |
|
Potasemia (mEq/l) |
4.92 |
4.5 |
4.65 |
4.42 |
4.14 |
4.04 |
|
Cloremia (mEq/l) |
106 |
109 |
106 |
104 |
102 |
102 |
|
Calcio (mg/dl) |
7.5 |
- |
- |
- |
- |
- |
|
Fósforo (mg/dl) |
3.1 |
- |
- |
- |
- |
- |
|
Magnesio (mg/dl) |
1.5 |
- |
- |
- |
- |
- |
|
VES (mm/1ºhora) |
5 |
2 |
2 |
- |
- |
- |
|
PCR (mg/L) |
40.4 |
26.8 |
32.5 |
- |
- |
- |
|
Procalcitonina ng/ml |
2.23 |
- |
3 |
- |
- |
- |
|
CPK (Ul/L) |
1635 |
- |
1395 |
973 |
- |
- |
|
LDH (Ul/L) |
1645 |
- |
1508 |
1557 |
- |
- |
|
TP (segundos) |
24.2 |
11.4 |
10.8 |
10.4 |
10.3 |
- |
|
KPTT (segundos) |
61.8 |
38 |
32 |
31 |
28 |
- |
|
pH |
7.36 |
- |
- |
- |
- |
7.34 |
|
pCO2 (mmHg) |
29. |
- |
- |
- |
- |
30.9 |
|
pO2 (mmHg) |
94.4 |
- |
- |
- |
- |
- |
|
EB (mEq/l) |
-8.5 |
- |
- |
- |
- |
-8.3 |
|
HCO3 (mEq/l) |
17.6 |
- |
- |
- |
- |
17.7 |
|
% Saturación |
96.7 |
- |
- |
- |
- |
- |
|
ProBNP (pg/ml) |
|
|
|
|
10724 |
|
> Orina completa: densidad 1006. pH 5. Proteinuria 0.17 g/l. Índice proteinuria creatininuria 1403. Glucosuria no detectable. Cetonuria no detectable. Hemoglobinuria 4+. Hematíes urinarios 4-5 por campo. Leucocitos 0-1 por campo. NaUr: 142. Urea Ur: 167. FeUrea: 55%.
> Orina de 24 horas: proteinuria 0.60 gr/24 hs. Creatininuria 374 mg/24 hs. Urea 5.39 g/24 hs. Sodio urinario 71.04 mEq/24 hs. Potasio 14.50 mEq/24 hs. Diuresis 740 ml. Clearance de creatinina 8 ml/min.
Sedimento urinario especializado: se observan cilindros granulosos, sin evidencia de eritrocitos dismórficos.
 |  |
> Frotis de sangre periférica: microhematocrito 22%. Avenocitos, hipocromía ++, microcitosis ++. Leucocitos 2000/mm3. Neutrófilos en cayado 4%. Neutrófilos segmentados 92%. Linfocitos 4%. Plaquetas 100000, macroplaquetas.
> Ecografía abdómino-renal: hígado: forma, tamaño y ecoestructura conservada. Parénquima homogéneo. Sin imagen de lesión focal. Vesícula: parcialmente colapsada por falta de ayuno. Vía biliar: de calibre conservado. Páncreas: forma, tamaño, contornos y ecoestructura conservadas. No se visualiza dilatación del conducto de Wirsung. Bazo: sin esplenomegalia. Parénquima homogéneo. Riñón derecho: situación normal, tamaño normal, morfología normal. Aumento de la ecogenicidad difusa. Vía excretora de calibre conservado. No se observan imágenes de litiasis. Riñón izquierdo: situación normal, tamaño normal, morfología normal. Aumento de la ecogenicidad difusa. Vía excretora de calibre conservado. No se observan imágenes de litiasis.
> Ecocardiograma: fracción de eyección: 71%. Diámetros de ventrículo izquierdo normales. Espesores parietales normales. Motilidad y engrosamiento sistólico normal. FSVI normal. Diámetros de aurícula izquierda normales. Raíz aórtica de diámetros normales. Morfología valvular normal. Cavidades derechas de diámetros normales. Pericardio con derrame leve circunferencial.
Radiografía de tórax: radiografía rotada. Índice cardiotorácico conservado. Radiopacidades heterogéneas parahiliares bilaterales, de límites mal definidos. Borramiento de ambos senos costofrénicos
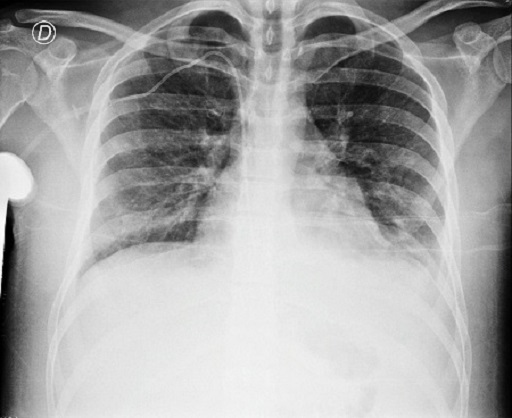
Tomografía de tórax con contraste: estudio artefactado. Estructuras ganglionares axilares bilaterales aumentadas en número, de rango no megálico. No se observan adenomegalias mediastinales. Se observan múltiples opacidades parcheadas en vidrio esmerilado de morfología pseudonodular, que se distribuyen en forma aleatoria bilateral, predominantemente en ambos lóbulos superiores, de probable etiología inflamatorio-infecciosa. Derrame pleural bilateral leve-moderado.
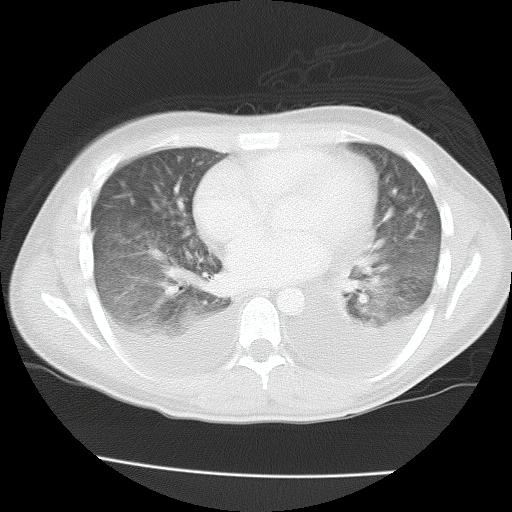
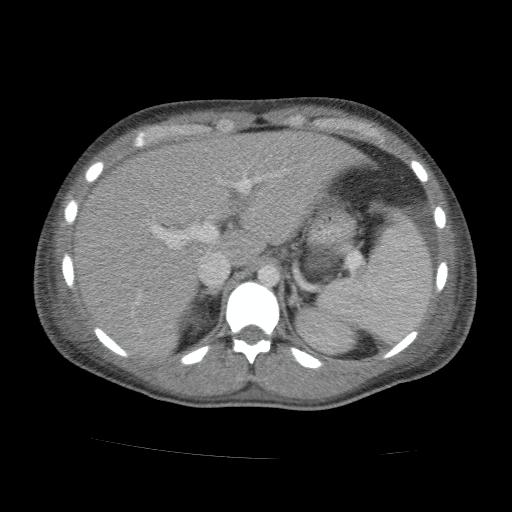
Tomografía de abdomen y pelvis con contraste: vesícula biliar colapsada. Esplenomegalia homogénea (DL 14 cm). Ambos riñones son de forma, tamaño y situación habituales. No concentran adecuadamente la sustancia de contraste. Sin evidencia de alteraciones calicopiélicas ni ureterales. Estructuras ganglionares inguinales bilaterales aumentadas en número, de rango no megálico. Escasa cantidad de líquido libre perihepático, periesplénico, ambas goteras parietocólicas y fondo de saco recto-vesical.
> Hemocultivos: negativos.
> Cultivo de punta de catéter: negativo.
> Laboratorio inmunológico:
- Fracción C3 complemento: 13 mg/dl.
- Fracción C4 complemento: 3 mg/dl.
> Serologías para enfermedades endémicas:
- Reacción de Huddleson: no reactivo.
- Reacción de Rosa de Bengala: no reactivo.
- Reacción de BPA: no reactivo.
- Leptospirosis (2º muestra): negativa.
- Fiebre Hemorrágica Argentina (2º muestra): negativo.
- Anticuerpos anti Trichinella: negativo.
> Cultivo de médula ósea bacteriológico y BAAR: negativo.
> Citometría de flujo de médula ósea: médula hipoplásica. Neutrófilos 80%. Eosinófilos 2.5%. Linfocitos 7%. Basófilos 4.5 %. 0.2 % serie nucleada. 4.5% serie displasia mieloeritroide. Serie roja displásica.
> Frotis de sangre periférica: microhematocrito 22%. Target cell. Dacrocitosis ++, avenocitos +++, esquistocitos ++. Leucocitos 4000/mm3. Neutrófilos en cayado 6%. Neutrófilos segmentados 78%. Eosinófilos 2%. Basófilos 0%. Linfocitos 2%. Monocitos 2%. 100000 macroplaquetas.
> Laboratorio inmunológico:
- Factor reumatoideo: 4 UI/ml.
- Fracción C3 complemento: 13 mg/dl.
- Fracción C4 complemento: 3 mg/dl.
- Anticuerpos anti ADN nativo: positivo 1/640.
> Perfil lipídico:
- Colesterol total: 114 mg/dl.
- Colesterol HDL 15 mg/dl.
- Colesterol LDL 23 mg/dl.
- Triglicéridos 505 mg/dl.
> Ferremia: 58 ug/dl.
> Ferritina: 1921.9 ng/ml.
*Los invitamos a dejar sus comentarios al pie del artículo. En dos semanas se publicará la segunda parte del caso clínico y, por último, la discusión final y conclusiones.
![]()