Aproximadamente 6.000 casos de leucemia linfoblástica aguda (LLA) son diagnosticados cada año en Estados Unidos; la mitad de los casos ocurren en niños y adolescentes. En los Estados Unidos, la LLA es el cáncer más común entre los niños y la causa más frecuente de muerte por cáncer antes de los 20 años de edad. Los síntomas de LLA presentes incluyen hematomas o sangrados por trombocitopenia, palidez y fatiga por anemia e infecciones causadas por neutropenia. La infiltración leucémica del hígado, el bazo, los ganglios linfáticos, y el mediastino es común al momento del diagnóstico.
La leucemia extramedular en el sistema nervioso central (SNC) o en los testículos puede requerir modificaciones específicas del tratamiento. Desde la primera descripción en 1948 de la remisión temporal de la leucemia inducida por la quimioterapia, la LLA pediátrica ha proporcionado un modelo para la mejora de la sobrevida entre los pacientes con cáncer mediante avances progresivos en la eficacia de los regímenes de quimioterapia y la estratificación de la intensidad del tratamiento de acuerdo con las características clínicas del paciente, las características biológicas de las células leucémicas, y la respuesta temprana al tratamiento, todos los cuales son predictivos del riesgo de recaída. Colectivamente, estos avances han aumentado la tasa de sobrevida de menos del 10% en la década de 1960 a 90% en la actualidad. Los nuevos descubrimientos revelan las promesas y desafíos de las estrategias de precisión médica que integran el genoma de la leucemia en el tratamiento contemporáneo.
Epidemiología y factores de riesgo
En los Estados Unidos, la incidencia de LLA es de unos 30 casos por millón de personas menores de 20 años de edad, con un pico de incidencia entre los 3 a 5 años de edad. La incidencia varía significativamente según la raza y el grupo étnico: 14,8 casos por millón de personas de raza negra, 35,6 casos por millón de personas de raza blanca y 40,9 casos por millón de hispanos. La LLA de la niñez se desarrolla más frecuentemente en niños que en niñas (relación varón: mujer, 55% a 45%).
Varios factores genéticos (principalmente el síndrome de Down) están asociados con un mayor riesgo de LLA, pero la mayoría de los pacientes no tienen factores heredados reconocidos. Los estudios de asociación genómica han identificado variantes polimórficas en varios genes (incluyendo ARID5B, CEBPE, GATA3, y IKZF1) que están asociadas con un aumento del riesgo de LLA o de subtipos de LLA específicos.
Determinadas mutaciones germinales raras en PAX5 y ETV6 están vinculadas a LLA familiar. Pocos factores de riesgo ambiental se asocian con LLA en niños. El aumento de las tasas de enfermedad se ha relacionado con la exposición a la radiación y a ciertos químicos, pero estas asociaciones explican sólo una muy pequeña minoría de los casos.
Bases genéticas de la LLA
La LLA comprende múltiples entidades con constelaciones distintas de alteraciones genéticas somáticas. Estas alteraciones genéticas incluyen aneuploidía (cambios en el número de cromosomas), reordenamientos cromosómicos que desregulan la expresión génica o resultan en la expresión de proteínas de fusión quimérica, deleciones y ganancias de ADN y mutaciones de la secuencia de ADN. En promedio, los genomas de la LLA infantil contienen sólo 10 a 20 mutaciones silenciosas no codificantes en el momento del diagnóstico y cerca del doble al momento de la recaída.
Muchas mutaciones perturban los procesos celulares clave, incluyendo la regulación transcripcional del desarrollo y la diferenciación linfoide; la regulación del ciclo celular; la vía de supresión tumoral de la proteína TP53 del retinoblastoma; señalización del receptor del factor de crecimiento, Ras, fosfatidilinositol 3-quinasa, y JAK-STAT; metabolismo nucleósido; y modificación epigenética. La perturbación de los últimos dos procesos es común en la recaída.
La LLA puede tener origen en precursores de células B o en el linaje de células T. En el 25 a 30% de los niños con LLA de células B, las células leucémicas tienen hiperdiploidía alta (> 50 cromosomas) debido a una ganancia cromosómica no aleatoria. Este subtipo se asocia con un excelente pronóstico. Se produce hipodiploidía (< 44 cromosomas) en 2 a 3% de los niños con LLA de células B, siendo un factor pronóstico negativo fuerte. La baja hipodiploidía (30 a 39 cromosomas), que se asocia con la presencia de mutaciones TP53 frecuentemente heredadas, es una manifestación del síndrome de Li-Fraumeni.
Las translocaciones cromosómicas y los reordenamientos intracromosómicos son eventos tempranos, posiblemente iniciales en la leucemia. Varios pueden ser detectados en muestras de sangre neonatal años antes de que existan manifestaciones clínicas de leucemia. Estas translocaciones y reordenamientos están generalmente presentes en todas las células leucémicas, se mantienen en la recaída, y con alteraciones genéticas adicionales, inducen leucemia en los sistemas de modelos experimentales.
Existen dos clases funcionales de translocaciones. La primera clase traslada oncogenes a las regiones reguladoras de genes transcriptos activamente, causando la expresión desregulada de una proteína intacta. Los ejemplos incluyen translocaciones que exponen el C-MYC al control de potenciadores de genes de la cadena pesada (IGH) o de la cadena liviana (IGK y IGL) de la inmunoglobulina en el linfoma y la leucemia de Burkitt, reordenamiento de los genes del factor 2 tipo receptor de citoquinas (CRLF2) y del receptor de eritropoyetina (EPOR) en IGH y IGK en la LLA de células B, y yuxtaposición de los factores de transcripción TLX1 y TLX3 al loci del receptor de células T (RCT) en la LLA de células T.
La segunda clase importante de translocaciones yuxtapone dos genes para codificar una proteína quimérica que tiene funciones distintas a la proteína de la cual deriva. Un ejemplo importante es la fusión ETV6-RUNX1, que fusiona dos factores de transcripción hematopoyéticos; se observa en una cuarta parte de los niños con LLA. Otros ejemplos importantes incluyen la translocación TCF3-PBX1, la translocación t (9; 22) (q34; q11.2) que resulta en la formación del cromosoma Philadelphia (Ph), y reordenamientos cromosómicos del gen de la leucemia de linaje mixto (LLM) que implican al cromosoma 11q23.
El cromosoma Ph codifica para BCRABL1, una tirosina quinasa activada. LLM (KMT2A) codifica una histona metiltransferasa que está involucrada en la regulación epigenética de las células sanguíneas en desarrollo. Más de 70 translocaciones diferentes se dirigen a la LLM, creando proteínas de fusión que median la autorrenovación aberrante de progenitores hematopoyéticos. Las translocaciones LLM son particularmente comunes en la LLA que se desarrolla antes del año de edad (75% de los casos). Las leucemias reorganizadas a LLM tienen muy pocas mutaciones somáticas adicionales, en particular en lactantes.
Estudios de secuenciación y de perfil genómico han identificado subtipos adicionales de LLA. Éstos incluyen casos con desregulación del factor de transcripción del gen ERG y casos con amplificación intracromosómica compleja del cromosoma 21.
En varios subtipos de LLA no hay una sola alteración cromosómica definida, Y estos subtipos se definen por otras características patológicas o genómicas. Por ejemplo, la LLA de células T precursoras temprana es una agresiva leucemia de stem cells y células progenitoras que tiene un inmunofenotipo distintivo y alteraciones genéticas dirigidas a factores de transcripción, vías de señalización y regulación epigenética. Los pacientes con LLA tipo Ph tienen un perfil de expresión génica de células leucémicas que es similar al de los pacientes con LLA Ph positivo, pero no tienen BCR-ABL1 y ocultan una amplia gama de alteraciones genéticas que activan la señalización de la tirosina quinasa.
Las alteraciones más comunes son fusiones que implican quinasas "clase ABL" (ABL1, ABL2, CSF1R, y PDGFRB), que pueden ser derivadas con inhibidores de ABL1 tales como imatinib y dasatinib, y fusiones, mutaciones o supresiones que activan la señalización JAK-STAT (incluyendo reordenamientos de JAK2, CRLF2, EPOR, y mutaciones de JAK1, JAK2, JAK3 y del receptor de la IL-7).
Con excepción de la leucemia con reordenamiento LLM en lactantes, cada uno de estos subtipos tiene típicamente múltiples alteraciones genéticas adicionales. Estas alteraciones habitualmente se dirigen a los genes que codifican proteínas implicadas en la señalización celular, la supresión tumoral y la diferenciación linfoide. Los dos genes diana más comunes que rigen el desarrollo linfoide B son PAX5 (mutado en el 35% de los casos de LLA en niños) y IKZF1 (mutado en el 15%).
Factores pronósticos
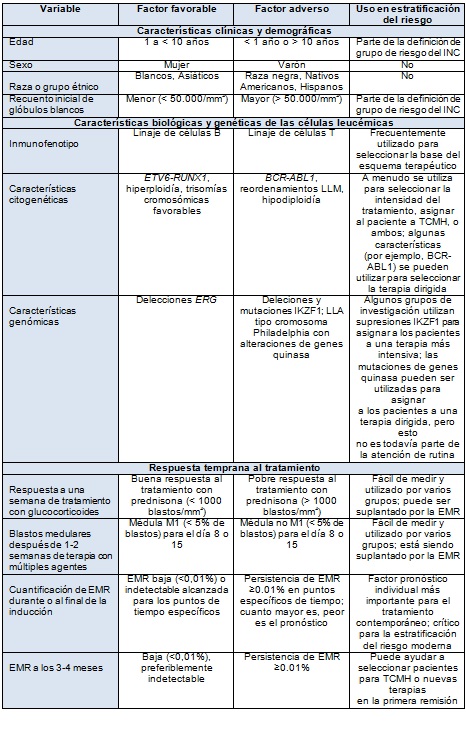
Los factores predictivos del aumento o la disminución de las posibilidades de curación son considerados cuando las decisiones se toman en base a la intensidad de la quimioterapia y la selección de los pacientes en primera remisión para el trasplante alogénico de células hematopoyéticas (Tabla 1). Los principales factores pronósticos incluyen las características clínicas que están presentes al momento del diagnóstico, las características biológicas y genéticas de las células leucémicas, y la respuesta temprana al tratamiento.
Características clínicas
La edad del paciente y el recuento inicial de glóbulos blancos son predictivos de los resultados, con la mayor edad o el mayor recuento de glóbulos blancos augurando un peor pronóstico. Una conferencia de consenso definió los subgrupos de LLA con "riesgo estándar" (edad de 1 a 9,99 años y recuento inicial de glóbulos blancos <50.000/mm3) y "alto riesgo" (edad ≥ 10 años, recuento inicial de glóbulos blancos ≥ 50,000/mm3, o ambos) comprendiendo, respectivamente, alrededor de dos tercios y un tercio de los niños con LLA de células B. Los niños menores de 1 año son un subgrupo especial de pacientes con peores resultados.
La edad y el recuento inicial de glóbulos blancos han limitado la importancia pronóstica en la LLA de células T. Varios subtipos de LLA ocurren con más frecuencia en ciertas razas y grupos étnicos, incluyendo LLA TCF3-PBX1 en la raza negra y LLA con reordenamiento CRLF2 en hispanos. Por lo tanto, las variaciones genéticas heredadas son importantes en la patogénesis de la LLA.
Inmunofenotipo
La expresión citoplasmática y en la superficie celular de los marcadores de linaje (inmunofenotipo) clasifica la LLA infantil en subgrupos de precursores de células B (85%) o de células T (15%) que son una reminiscencia de las etapas normales de la maduración linfoide. Los pacientes con leucemia o linfoma de Burkitt tienen un inmunofenotipo de células B maduras, con expresión de inmunoglobulina en la membrana celular, reordenamiento del oncogén MYC, y un curso clínico agresivo pero curable. Muchas mutaciones que están vinculadas a la leucemogénesis están dirigidas a genes que regulan la diferenciación normal de células B o de células T, deteniendo la diferenciación.
Los pacientes con LLA de células T a menudo son del sexo masculino, de raza negra, de mayor edad y con menor probabilidad de ser hispanos que los pacientes con LLA de células B, tienen recuentos de glóbulos blancos iniciales más altos que los pacientes con LLA de células B, y tienen compromiso de los ganglios linfáticos del mediastino y del SNC. Históricamente, la sobrevida de los niños con LLA de células T era inferior a la de los niños con LLA de células B. Con el uso de un tratamiento más intensivo, esta diferencia se ha reducido sustancialmente. Algo de la preponderancia de la LLA de células T entre los niños y hombres jóvenes puede deberse a mutaciones específicas dirigidas a los genes del cromosoma X.
Características genéticas y biológicas
Varias alteraciones genéticas se asocian con el resultado en niños con LLA. La alta hiperdiploidía y el t críptico (12; 21) que codifica para ETV6-RUNX1 se asocian con un resultado favorable. La hipodiploidía con menos de 44 cromosomas, el reordenamiento LLM, el BCR-ABL1, la LLA tipo Ph, el reordenamiento CRLF2, la amplificación intracromosómica del cromosoma 21, y la LLA de células T precursoras temprana se asocian con características clínicas de alto riesgo o un mal resultado.
Las alteraciones de IKZF1, que codifica para el factor de transcripción linfoide Ikaros, son comunes en la LLA Ph-positivo y en la LLA tipo Ph. Estas alteraciones también se asocian con un pobre resultado.
Respuesta temprana al tratamiento
El tiempo necesario para eliminar la carga de población de células leucémicas a niveles indetectables es el factor pronóstico más poderoso en la LLA infantil. Los niveles submicroscópicos de enfermedad mínima residual en la LLA (1 célula leucémica por 104 a 105 células normales) se pueden medir mediante amplificación por reacción en cadena de la polimerasa de los reordenamientos de genes IGH o TCR clonotípicos que son únicos de la leucemia de un paciente individual o por detección mediante citometría de flujo de combinaciones aberrantes de antígenos en la superficie celular.
El riesgo de fracaso del tratamiento y de muerte es 3 a 5 veces más alto entre los niños con niveles de enfermedad mínima residual del 0,01% o mayores al final de la terapia de inducción y en puntos de tiempo posteriores que entre aquellos con niveles inferiores al 0,01%. La intensificación del tratamiento para los pacientes con niveles más altos de enfermedad mínima residual mejora los resultados. Las técnicas de secuenciación de próxima generación emergentes para la detección de enfermedad mínima residual pueden ser útiles al proporcionar una detección sensible de células leucémicas por debajo del nivel detectado con otras técnicas.
Tratamiento
Mejoras en la sobrevida con el tiempo
Hace casi 50 años, la quimioterapia de combinación inducía a la remisión (desaparición de la evidencia clínica de leucemia y restauración de la hematopoyesis normal) en el 80 a 90% de los niños con LLA. Sin embargo, la enfermedad recayó en casi todos estos niños, por lo general en el SNC, con tasas de sobrevida del 10 a 20%. La sobrevida aumentó considerablemente con la adición de la irradiación craneoespinal o craneal y de la quimioterapia intratecal.
Un hito importante en el tratamiento de niños con LLA fue el desarrollo de un régimen intensivo de inducción y consolidación de 8 semanas y con 8 drogas introducido por Riehm y col. Este régimen, que ahora se llama protocolo I, se convirtió en la base para el régimen Berlín-Frankfurt-Münster, que es el núcleo de la mayoría de las terapias actuales para LLA.
Desde que se introdujo este régimen, grandes grupos de investigación cooperativa e instituciones individuales han enrolado al 75-95% de los niños con diagnóstico de LLA de América del Norte y Europa Occidental en ensayos clínicos. Estos ensayos han dado lugar a mejoras notables en la sobrevida, con tasas de sobrevida libre de eventos a cinco años de hasta el 85% y tasas de sobrevida global de hasta el 90%, según los datos más recientemente reportados.
Tratamiento contemporáneo
Los componentes básicos de las diversas terapias para los niños con LLA son similares e incluyen varias fases discretas. La terapia de inducción tiene una duración de 4 a 6 semanas e incluye una semana de glucocorticoides (prednisona o dexametasona), vincristina, una preparación de asparaginasa, el uso opcional de una antraciclina y quimioterapia intratecal. Casi todos los pacientes alcanzan la remisión, pero esto no es una cura, ya que la recaída ocurrirá universalmente sin terapia adicional.
Después de la remisión, el tratamiento incluye 6 a 8 meses de quimioterapia de combinación intensiva que está diseñada para consolidar la remisión y prevenir el desarrollo de leucemia manifiesta en el SNC. Luego se administra tratamiento como una fase de intensificación retardada de 8 semanas (Protocolo II), en base al protocolo I Berlín-Frankfurt-Münster. Los cursos repetidos de metotrexate, administrado ya sea a través de infusión intravenosa corta o en altas dosis durante 24 horas, seguido de la administración de ácido folínico para "rescatar" los tejidos normales de los efectos tóxicos, son un componente crítico de todos los regímenes contemporáneos para LLA.
Los pacientes reciben entonces terapia de mantenimiento de baja intensidad basada en "antimetabolitos" durante 18 a 30 meses. Esta terapia consiste en mercaptopurina o tioguanina oral diaria y metotrexate oral semanal. Algunos regímenes también incluyen pulsos periódicos por 5 a 7 días de glucocorticoides y vincristina. Las razones exactas por las que se requiere una terapia de mantenimiento y la composición y la duración más eficaces de la quimioterapia se desconocen.
Debido a que el tratamiento de mantenimiento es prolongado y a que requiere la administración diaria de un fármaco oral, la adhesión puede ser problemática; 20% de los pacientes tienen una adherencia menor al 90%, y la disminución de la adherencia se asocia con un riesgo de recaída que es 4 veces más alto que el riesgo entre los pacientes cuya tasa de adherencia es del 90% o mayor. Los polimorfismos del huésped pueden influenciar tanto la eficacia como la toxicidad de la mercaptopurina, que es la columna vertebral de la terapia de mantenimiento.
Terapia dirigida al SNC
La irradiación craneal mejoró dramáticamente las tasas de curación de los pacientes con LLA en las décadas de 1960 y 1970, pero se asoció con un aumento del riesgo de tumores secundarios del SNC, retraso en el crecimiento, endocrinopatías y alteraciones neurocognitivas. En consecuencia, la irradiación del SNC se ha limitado a subgrupos de pacientes cada vez más pequeños a través del tiempo.
Varios grupos de investigación han eliminado la irradiación del SNC para la mayoría o todos los niños con LLA de reciente diagnóstico, y sus resultados son bastante similares a los obtenidos por los grupos que siguen incluyendo la irradiación en la terapia para niños con LLA. El rol actual de la irradiación del SNC es controversial, pero todos los grupos ahora tratan al menos al 80% de los niños que han sido recién diagnosticados sin el uso de la irradiación craneal.
Tratamiento de la LLA recaída, incluyendo el Trasplante de Células Hematopoyéticas (TCH)
La recaída se produce en el 15 a 20% de los niños con LLA, y las tasas de curación son mucho más bajas después de la recaída. Los factores pronósticos en la recaída incluyen el tiempo hasta la recaída (un tiempo más corto se asocia con un peor pronóstico), el inmunofenotipo (el inmunofenotipo de células T se asocia con un peor pronóstico), y el sitio de recaída (la enfermedad de la médula ósea se asocia con peor pronóstico que la enfermedad extramedular).
Las células leucémicas obtenidas de pacientes en recaída temprana frecuentemente albergan mutaciones que disminuyen la sensibilidad a los fármacos quimioterápicos comunes. Si la recaída se produce después de terminar el tratamiento primario, la mayoría de los niños entrarán en una segunda remisión, y la posibilidad de curación es de aproximadamente 50%. Si la recaída se produce durante la terapia, la posibilidad de alcanzar una segunda remisión es sólo del 50 al 70%, y sólo 20 a 30% de los pacientes se curan.
El trasplante alogénico de células hematopoyéticas se utiliza mucho más comúnmente después de la recaída (en ≥ 50% de los pacientes) que durante el tratamiento primario (5 al 10% de los pacientes). La evaluación de la respuesta de la enfermedad mínima residual puede ser útil para determinar cuáles pacientes deben ser sometidos a trasplante durante una segunda remisión y cuáles pacientes no.
La LLA es frecuentemente una enfermedad policlonal, y mutaciones en subclones pueden ser seleccionadas por la quimioterapia y promover la resistencia. Éstas incluyen mutaciones CREBBP que están vinculadas con resistencia a los glucocorticoides y mutaciones NT5C2 y PRPS1 que están asociadas con resistencia a las tiopurinas. En futuros estudios, será importante identificar las mutaciones emergentes que están asociadas con resistencia y explorar el potencial para la modificación de la terapia a fin de eludir la recaída.
Terapia dirigida y medicina de precisión
Las mejoras dramáticas en la sobrevida de los niños con LLA en los últimos 50 años se deben casi exclusivamente a la identificación de dosis y esquemas de agentes quimioterápicos más eficaces que han estado ampliamente disponibles desde hace décadas más que al desarrollo de nuevos tratamientos. Los recientes descubrimientos en relación a las bases genéticas de la LLA y el desarrollo de terapias dirigidas a las lesiones moleculares que afectan a la sobrevida de las células leucémicas han allanado el camino para el uso expansivo de enfoques de medicina de precisión para el tratamiento del cáncer.
Un ejemplo notable es el uso de inhibidores de tirosina quinasa en pacientes con leucemia mieloide crónica, un cáncer que es accionado por la oncoproteína de fusión BCR-ABL1. El tratamiento con inhibidores de tirosina quinasa (imatinib y agentes relacionados) ha convertido a la leucemia mieloide crónica de una enfermedad con requerimiento de terapia intensiva y a menudo de TCH a una enfermedad crónica que puede en la mayoría de los casos ser manejada con éxito durante décadas con inhibidores de tirosina quinasa orales, con el potencial para la interrupción del tratamiento en algunos pacientes.
La proteína de fusión BCR-ABL1 también se produce en el 25% de los adultos y en el 3 a 5% de los niños con LLA (LLA Ph-positivo), y en la LLA, en comparación con la leucemia mieloide crónica, se asocia con alteraciones genéticas secundarias, en particular alteraciones del IKZF1. Antes del uso de inhibidores de la tirosina quinasa, menos de la mitad de los niños con LLA Ph-positivo sobrevivían. La combinación de imatinib con quimioterapia citotóxica ha demostrado ser altamente eficaz en niños con LLA Ph-positivo y ha reducido al mínimo la necesidad de TCH en la primera remisión.
La LLA tipo Ph se asocia con un pobre pronóstico, y es una candidata lógica para la terapia con inhibidores de la tirosina quinasa adaptada individualmente. Una amplia gama de alteraciones genéticas activa la señalización quinasa en la LLA tipo Ph; éstas incluyen una alta frecuencia de reordenamientos que convergen en un número limitado de vías de señalización, incluyendo la señalización clase ABL y JAK-STAT.
Grandes estudios preclínicos demuestran que la activación de las vías de señalización inducida por estas alteraciones es sensible a los inhibidores de tirosina quinasa; esto sugiere que los enfoques de medicina de precisión deberían ser exitosos en todos los subgrupos de LLA. Estos hallazgos son apoyados por informes anecdóticos de las respuestas dramáticas de la LLA tipo Ph refractaria a quimioterapia a la terapia con inhibidores de tirosina quinasa. Esto es especialmente importante en niños mayores y adultos, en los que la LLA tipo Ph es más común.
Un reto importante en el diseño de futuros ensayos clínicos será garantizar el enrolamiento adecuado de los pacientes que albergan cada clase de alteración genética. Para hacer frente a este desafío, tendrán que desarrollarse ensayos clínicos internacionales que involucren múltiples grupos cooperativos, como se ha realizado con éxito en estudios de LLA Ph-positivo.
Inmunoterapia
CD19 es un antígeno de la superficie celular que está presente en alta densidad en la mayoría de las células leucémicas tipo B. Varios grupos han desarrollado estrategias para transducir células T autólogas con un fragmento de anticuerpo anti-CD19 acoplado a los dominios intracelulares de señalización del receptor de células T, lo que permite redirigir a los linfocitos T citotóxicos para que reconozcan y eliminen a las células leucémicas tipo B. Estas células T proporcionan una opción terapéutica nueva importante.
En un estudio, 30 niños con LLA tratada intensivamente y con múltiples recaídas fueron tratados con estas células T quiméricas con receptor de antígeno modificado; el 90% de los niños alcanzó la remisión, con remisión sostenida en alrededor de dos tercios de los mismos. Aproximadamente tres cuartas partes de los niños estaban vivos 6 meses después de la infusión. Las remisiones fueron duraderas, con 1 a 3 años de seguimiento. Muchos pacientes presentaron un síndrome de liberación de citoquinas grave después de la activación de las células T citotóxicas in vivo. Este síndrome fue acompañado por altos niveles de IL-6 en suero que podrían ser tratados con éxito con el anticuerpo anti-IL-6 tocilizumab. Están en curso estudios de durabilidad de la terapia de receptor de antígeno quimérico de células T y su rol en los pacientes con LLA con enfermedad menos avanzada.
Una estrategia diferente para aprovechar la respuesta inmune de las células T contra las células leucémicas es proporcionada por el blinatumomab, un anticuerpo modificado genéticamente que contiene fragmentos que reconocen tanto el CD19 como el CD3 (que está presente en todas las células T) y que por lo tanto pone a las células T en contacto directo con las células leucémicas B, permitiendo que las células T citotóxicas las eliminen. El blinatumomab actualmente está siendo evaluado en niños con una primera recaída de LLA de células B.
Efectos tóxicos del tratamiento a corto plazo y largo plazo
Aproximadamente del 1 al 2% de los niños con LLA mueren antes de alcanzar la remisión, y un adicional de 1 a 2% mueren por los efectos tóxicos durante la remisión. Los pacientes con síndrome de Down, los lactantes, los adolescentes mayores, y aquellos que reciben terapia más intensiva tienen un mayor riesgo de muerte por efectos tóxicos, principalmente debido a infección. Los riesgos pueden ser mitigados mediante modificaciones en la terapia y el tratamiento de apoyo. A medida que las tasas de curación para la LLA infantil mejoran, la muerte relacionada con el tratamiento representa un porcentaje más alto de todas las muertes.
Uno de los problemas más acuciantes asociados con la terapia actual para la LLA es la osteonecrosis, que ocurre en el 5 a 10% de los pacientes. El riesgo es mucho mayor entre los adolescentes (15 a 20%) que entre los niños pequeños, y las niñas son afectadas con mayor frecuencia que los varones. La osteonecrosis más comúnmente afecta a grandes articulaciones, en especial las caderas, las rodillas, los hombros y los tobillos, y a menudo requiere tratamiento quirúrgico, incluyendo reemplazo de la articulación. Las modificaciones en los esquemas de administración de glucocorticoides pueden disminuir el riesgo de osteonecrosis.
Efectos adicionales relacionados con el tratamiento incluyen síndrome metabólico, obesidad, alteraciones cardiovasculares y efectos tóxicos del SNC y periférico. Estos efectos son causados por agentes antileucémicos muy eficaces, y el riesgo de efectos tóxicos en una persona se ve influenciado por factores genéticos del huésped y por la actividad y metabolismo del fármaco involucrado. Por lo tanto, un objetivo importante es la adaptación de la exposición al fármaco de acuerdo con el riesgo previsto de recaída y de efectos tóxicos específicos.
Un niño curado de LLA se espera que tenga entre 60 y 80 años de vida útil restante. Las preguntas críticas son si esa expectativa de vida se ve acortada por la leucemia, su tratamiento, o ambos, si ciertas condiciones de salud crónicas que afectan la vida diaria se desarrollan con una frecuencia más alta o con mayor gravedad en los sobrevivientes que en las personas que nunca recibieron tratamiento para la LLA infantil, y si persisten efectos emocionales o neurocognitivos duraderos que limitan la plena realización del potencial del paciente.
Desafortunadamente, muchos sobrevivientes de LLA tienen efectos tóxicos crónicos, y las alteraciones neurocognitivas parecen aumentar a medida que se acercan a la edad media. El seguimiento continuo a largo plazo de las personas que tuvieron LLA en la infancia es esencial para definir los riesgos y desarrollar estrategias para disminuirlos, así como para disminuir los efectos tóxicos del tratamiento.
Implicaciones del éxito del tratamiento
Las tasas de sobrevida de los adolescentes con LLA son inferiores a las de los niños pequeños, y la sobrevida es aún peor entre los adultos jóvenes. Las razones de estas diferencias son multifactoriales e incluyen factores del tratamiento, una mayor prevalencia de subtipos genéticos desfavorables entre los pacientes de mayor edad, la disminución de la capacidad de los adolescentes y adultos jóvenes de recibir tratamiento intensivo sin presentar efectos adversos secundarios, y factores sociales tales como la cobertura de los seguros y la falta de supervisión parental de la terapia. Las instituciones y los grupos cooperativos que tratan a adultos jóvenes con LLA han adoptado con éxito un tratamiento basado en regímenes pediátricos. Esta estrategia es factible para pacientes de hasta alrededor de 50 años de edad, con los primeros resultados sugiriendo importantes mejoras en la sobrevida.
Dado que la población de niños es mayor en países de medianos y bajos ingresos que en los países de altos ingresos, el número total de niños con diagnóstico de LLA es también mayor en estos países; estos niños tienen una sobrevida inferior en comparación con los niños tratados en países de altos ingresos. Dado que la LLA puede ser diagnosticada con técnicas sencillas y tratada exitosamente con agentes quimioterapéuticos relativamente baratos, es factible mejorar rápidamente el resultado de los niños con LLA en los países de bajos y medianos ingresos. Las asociaciones y relaciones entre los centros de altos ingresos de América del Norte y Europa occidental y los centros de cáncer pediátrico de Asia, América Central y América del Sur, y Europa del Este han mejorado sustancialmente la sobrevida de los niños con LLA.
Conclusiones
En los últimos años se observaron avances tremendos en la comprensión de la biología de la LLA y una notable eficacia de los enfoques terapéuticos biológicos y químicos dirigidos para una enfermedad de lo contrario refractaria. Se prevé que en los próximos años el paisaje genómico de la LLA será descripto completamente, las causas biológicas del fracaso del tratamiento totalmente dilucidadas, y los roles de una amplia gama de nuevos productos químicos y biológicos definidos. A medida que la tasa de curación de la LLA infantil se acerque al 100%, los principales retos serán identificar a las personas que requieren una terapia menos intensiva para alcanzar la curación y purificar los regímenes tóxicos y complejos para incorporar enfoques más simples y seguros que darán como resultado una alta calidad de vida, junto con una mayor sobrevida a largo plazo.
(Ver Tabla 1 a continuación)
Comentario: La leucemia linfoblástica aguda es la principal causa de muerte por cáncer en la población pediátrica; la edad, ciertos factores genéticos y ambientales, la respuesta al tratamiento y a los efectos tóxicos del mismo son determinantes importantes en la evolución y la sobrevida de estos pacientes. Los avances en el diagnóstico y manejo de la LLA han mejorado notablemente la capacidad de respuesta de estos pacientes; el desafío a futuro será diseñar esquemas de tratamiento que sean altamente eficaces pero con menor toxicidad, a fin de mejorar la calidad de vida del paciente mientras se logra la remisión, y evitar efectos nocivos a largo plazo que afecten la sobrevida.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol