Introducción
La trombofilia es una anomalía de la coagulación que aumenta el riesgo de trombosis. Puede ser hereditaria o adquirida, aunque tanto factores genéticos como ambientales influyen sobre la coagulación y pueden interactuar para provocar episodios trombóticos.
¿Cuáles son los tipos de trombofilias hereditarias?
En la práctica clínica los factores hereditarios más evaluados que predisponen a la trombosis venosa son: deficiencias genéticas de los anticoagulantes naturales (antitrombina, proteína C y proteína S) y polimorfismos genéticos “de ganancia de función” (factor V de Leiden y mutación del gen de protrombina). Estas dos últimas trombofilias identificadas recientemente son frecuentes en la población general. Estudios sugirieron que las trombofilias hereditarias son factores pronósticos modestos de un primer episodio de tromboembolia venosa (TEV) y que su importancia es limitada para pronosticar episodios recidivantes.
¿Son frecuentes las trombofilias hereditarias? , ¿cómo afectan el riesgo de trombosis?
La mayor parte de las trombofilias hereditarias son defectos heterocigotos. La tabla resume su frecuencia y sus riesgos relativos.
Tabla - Riesgos relativos y frecuencias aproximadas de las diferentes trombofilias hereditarias en la población del Reino Unido
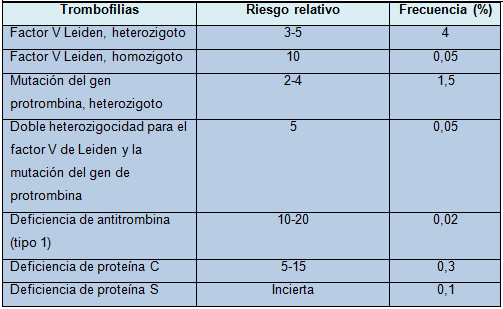
Polimorfismos de ganancia de función
Factor V de Leiden
La trombofilia más frecuente es el factor V de Leiden. Su frecuencia varía considerablemente según el origen étnico. En las poblaciones blancas de origen europeo, la frecuencia es de aproximadamente el 3-7%, similar a la observada en poblaciones del Mediterráneo y de Medio Oriente. En cambio el factor V de Leiden no se encuentra en el sudeste asiático, Japón y el África subsahariana ni en las poblaciones indígenas de Australia.
El polimorfismo del factor V de Leiden es una mutación de sentido erróneo. El resultado de esta mutación es que el factor V activado, un factor de coagulación, se inactiva unas 10 veces más lentamente que lo normal por la proteína C activada y esto lleva al aumento de la generación de trombina. La base molecular de esta resistencia a la proteína C activada fue identificada inicialmente por investigadores de la Universidad de Leiden en los Países Bajos.
En relación con no portadores, los portadores heterocigotos del factor V de Leiden tienen de tres a cinco veces más riesgo de TEV durante su vida. El riesgo absoluto, sin embargo, es bajo, ya que sólo el 5% de estas personas ha sufrido un episodio de TEV al llegar a los 65 años. Aproximadamente el 20% de los pacientes con un primer episodio no provocado de TEV son portadores heterocigotos de factor V de Leiden. Por razones inexplicadas, este factor parece ser un factor de riesgo más fuerte para las trombosis venosas profundas que para la embolia pulmonar. Aquéllos que son homocigotos para el factor V de Leiden tienen 10 veces o más riesgo de TEV que los no portadores. Sin embargo, como son poco frecuentes en la población, no contribuyen sustancialmente a la carga global de TEV.
Se hallaron interacciones con otros factores de riesgo para VTE, en especial los anticonceptivos orales combinados, el tratamiento de reemplazo hormonal y el embarazo. Sin embargo, el riesgo absoluto de TEV mientras se toman anticonceptivos orales combinados en el 5% de la población que es portador del gen del factor V de Leiden sigue siendo demasiado pequeño como para justificar la pesquisa sistemática.
Mutación del gen de protrombina
La mutación del gen de protrombina es la segunda trombofilia hereditaria más frecuente, siendo alrededor del 1-2% de las poblaciones blancas de origen europeo portadores heterocigotos. La mutación se asocia con dos a cuatro veces mayor riesgo de TEV durante toda la vida. Se la halla en alrededor del 5% de personas con el primer episodio no provocado de TEV.
Las cifras de protrombina están aumentadas en un 30% en heterocigotos y un 70% en homocigotos.
Deficiencias de los anticoagulantes naturales
Deficiencia de antitrombina
Las mutaciones heterocigotas del gen para antitrombina, antes llamado antitrombina III, producen deficiencia de antitrombina que lleva a menor inhibición del factor Xa y de la trombina y por lo tanto al aumento de la generación y de la actividad de la trombina. Los dos tipos principales de deficiencia de antitrombina son:
- tipo I (defecto cuantitativo con disminución de la producción de una molécula normal)
- tipo II (defecto cualitativo en el cual la molécula en sí es anormal).
La deficiencia de antitrombina tipo II se puede subdividir según el lugar del defecto molecular: lugar de fijación de la heparina (que tiene menor riesgo de trombosis), lugar reactivo o ambos. En la práctica es difícil distinguir entre los subtipos y se debe efectuar la interconsulta con un especialista.
La deficiencia de antitrombina tipo I es rara (0,02% de la población) y las mutaciones homocigotas son incompatibles con la vida. El grado de aumento de riesgo de TEV asociado con la deficiencia de antitrombina heterocigota sigue siendo discutido. Diferentes diseños de estudios llegaron a conclusiones diferentes, con riesgos relativos estimados desde similares a los del factor V de Leiden a riesgos cinco veces mayores. Algunos aconsejan emplear un concentrado de antitrombina en ciertos pacientes de alto riesgo con deficiencia de antitrombina y TEV previo sometidos a procedimientos donde se debe suspender la anticoagulación. Estas situaciones son raras y se debe efectuar la interconsulta con un especialista.
Deficiencia de proteína C y proteína S
La proteína C y la proteína S son glucoproteínas dependientes de la vitamina K. La deficiencia hereditaria de cualquiera de ellas disminuye la inactivación del factor Va y el VIIIa y por lo tanto aumenta la generación de trombina. La deficiencia de proteína C se clasifica en tipo I (cuantitativa) y tipo II (cualitativa), aunque a diferencia de la deficiencia de antitrombina el riesgo de trombosis entre los diferentes tipos es similar. Igualmente, existe la deficiencia tipo I y tipo II caracterizadas por cifras normales de proteína S, pero cifras disminuidas de proteína activa libre. Se mencionó una deficiencia tipo III caracterizada por cifras normales de proteína S total, pero cifras reducidas de las proteína activa libre, que probablemente representa producción deficiente (deficiencia tipo I) no reconocida con la prueba menos sensible para proteína S total.
Alrededor del 0,3% de la población tiene deficiencia de proteína C. Las estimaciones de riesgo de TEV asociado con deficiencia de proteína C son muy variables. A diferencia de la deficiencia de antitrombina, la deficiencia homocigota de proteína C, aunque rara, es compatible con la vida y se manifiesta con púrpura neonatal fulminante o trombosis de las venas cerebrales.
La frecuencia de deficiencia de proteína S es probablemente de alrededor del 0,1%. El riesgo de TEV asociada con esta deficiencia es incierto: estudios de familias sugieren que el riesgo es similar al observado en la deficiencia de proteína C, pero estudios demográficos sugieren un riesgo menor.
Doble heterocigosidad
Es posible heredar más de una tendencia trombofílica. Esto se ve con más frecuencia en aquéllos que son heterocigotos para el factor V de Leiden y la mutación del gen de protrombina. Aunque estas personas pueden tener mayor riesgo de TEV que las que son heterocigotas para un solo gen, un metanálisis reciente halló que el riesgo es similar al del factor V de Leiden solo.
¿Cuál es la importancia de los antecedentes familiares?
Cuando se estiman los riesgos asociados con las trombofilias hereditarias en pacientes asintomáticos es importante saber que el riesgo absoluto depende no sólo de la trombofilia identificada, sino también de si el paciente tiene antecedentes familiares de TEV, lo que es en sí mismo un factor de riesgo para TEV. En familias con trombofilia, aquéllos sin trombofilia familiar aún tienen mayor riesgo de TEV que la población general. Esto sugiere la presencia de otros factores de riesgo hereditarios o adquiridos en estas familias aunque en la actualidad no se los pueda identificar.
¿Cómo se diagnostican las trombofilias hereditarias?
Los estudios para la trombofilia hereditaria incluyen una cantidad de pruebas de coagulación complejas junto con pruebas genéticas (cuadro)
|
Pruebas estándar para las trombofilias hereditarias y causas adquiridas frecuentes de alteraciones Tiempo de protrombina |
Interpretar los resultados puede ser difícil. Se deben evitar los estudios para trombofilia durante la fase aguda de un episodio trombótico, ya que varios factores pueden influir sobre los resultados y la presencia de una trombofilia hereditaria no afecta el tratamiento inicial de la TEV. Es importante saber qué medicamentos recibe el paciente. La heparina puede disminuir los valores de antitrombina y la warfarina disminuirá los valores de las proteínas dependientes de la vitamina K, como la proteína C y la proteína S. Además, las cifras de proteína S descienden con los medicamentos que contienen estrógenos. El estado de salud de los pacientes también puede afectar las pruebas de coagulación. En la coagulación intravascular diseminada y en las enfermedades hepáticas los valores de los anticoagulantes naturales disminuyen. En el embarazo, la proteína S desciende debido a los estrógenos. La antitrombina puede disminuir en el síndrome nefrótico debido a la albuminuria. Los neonatos tienen valores bajos de antitrombina, proteína C y proteína S, que en general llegan a los valores adultos a los 3-6 meses.
La pesquisa para las trombofilias hereditarias incluye la medición de antitrombina, proteína C, proteína S y pruebas para el factor V de Leiden y las mutaciones del gen de protrombina. El tiempo de protrombina y el tiempo de tromboplastina parcial activada ayudan a identificar a los pacientes que reciben anticoagulantes y por ende a interpretar los resultados.
Se debe señalar que la mayoría de los pacientes con valores por debajo del límite inferior tradicional (valores inferiores a dos desviaciones estándar por debajo de la media, es decir 1 en 40 pacientes) no tendrán deficiencia hereditaria de los anticoagulantes naturales.
¿Cuándo se debería investigar en busca de trombofilias hereditarias?
Tromboembolia venosa
Los médicos pueden querer pesquisar la trombofilia hereditaria en los pacientes que sufren TEV y sus familiares asintomáticos por dos motivos. Primero, para ofrecer anticoagulación prolongada al probando inicial. Segundo, para identificar a familiares asintomáticos con riesgo aumentado de TEV a fin de que una intervención no adecuada para todos los familiares se pueda emplear para disminuir su riesgo individual de TEV. Sin embargo, ¿Se justifican estas consideraciones?
Tratamiento del probando inicial
Factores de riesgo de recidiva
Para el probando inicial se pronostica aumento del riesgo de recidiva ante un factor de riesgo persistente (por ejemplo, cáncer) o un episodio no provocado. El riesgo de recidiva aumenta tras un primer episodio no provocado de TEV si el paciente es varón o los valores de D-dímero están aumentados tras finalizar la anticoagulación. En estos casos es conveniente la anticoagulación prolongada para la prevención secundaria. Los antecedentes familiares de trombosis no aumentan el riesgo de recidiva. Estudios de cohortes prospectivos mostraron que la trombofilia hereditaria no es factor pronóstico de recidiva. Una revisión sistemática de las dos trombofilias hereditarias halló que el aumento de riesgo de recidiva no es de magnitud suficiente para justificar la anticoagulación prolongada. Los pacientes con defectos combinados (pacientes homocigotos para el factor V de Leiden o la mutación del gen de protrombina o dobles portadores heterocigotos de factor V de Leiden y la mutación del gen de protrombina ) no parecen tener alto riesgo de recidiva. Además, las trombofilias hereditarias no son factores pronósticos de riesgo del síndrome postrombótico tras una trombosis venosa profunda ni tampoco de mortalidad.
Duración de la anticoagulación
Las trombofilias hereditarias parecerían tener sólo un papel secundario en la duración del tratamiento anticoagulante. Recomendaciones británicas recientes sugieren que sólo se deben estudiar los pacientes con episodios no provocados y antecedentes familiares de trombosis que planean suspender la anticoagulación.
La situación es distinta en las trombosis venosas en ubicaciones poco frecuentes. Aquí hay menos datos aún y aunque algunos sugieren que la presencia de trombofilia hereditaria debería dar lugar a tratamiento anticoagulante más prolongado, no hay buena evidencia para estas recomendaciones.
Embarazo y anticoncepción
Los anticonceptivos orales combinados están contraindicados en mujeres con un episodio previo de TEV y esto no cambia con la presencia o la ausencia de una trombofilia hereditaria. No obstante, el dispositivo intrauterino liberador de levonorgestrel y los comprimidos con dosis bajas de progestágeno parecen ser seguros. En el embarazo, todas las mujeres con un episodio previo de TEV deberán recibir tromboprofilaxis durante las primeras seis semanas posparto. También se deberá ofrecer tromboprofilaxis prenatal a las mujeres con antecedentes de TEV no provocado o provocado por estrógenos. Aquéllas con un episodio previo provocado por un factor de riesgo transitorio menor no deben recibir sistemáticamente tromboprofilaxis prenatal, pero se les debe efectuar estudios para la tendencia trombofílica, ya que un resultado positivo podría hacer cambiar esta decisión. La presencia o la ausencia de una trombofilia hereditaria no afecta la dosis de heparina de bajo peso molecular empleada para la tromboprofilaxis en el embarazo, salvo en mujeres con deficiencia de antitrombina, donde las recomendaciones del Royal College of Obstetricians and Gynaecologists (Reino Unido, UK) aconsejan dosis más altas), aunque las recomendaciones de los EEUU no hacen esa distinción.
Tratamiento de los familiares asintomáticos
Puesto que la anticoagulación prolongada de los familiares asintomáticos con trombofilia no se recomienda, sólo preocupa la posibilidad de tratar de prevenir episodios no provocados de TEV en los familiares. Los estudios indican que los familiares con trombofilia podrían recibir tromboprofilaxis o profilaxis más prolongada en situaciones de alto riesgo, pero no estaría indicada para familiares sin trombofilia. Los antecedentes familiares de TEV son de por sí un factor de riesgo de TEV, aunque no se hallen trombofilias hereditarias. Por lo tanto parece razonable ofrecer tromboprofilaxis en situaciones de alto riesgo a todos los familiares de primer grado de pacientes con TEV aunque el resultado de las pruebas de detección de trombofilia sea negativo. Puesto que hallar una trombofilia hereditaria tiene poco impacto sobre el tratamiento de los familiares asintomáticos, las recomendaciones del National Institute for Health and Care Excellence aconsejan no ofrecer sistemáticamente pruebas de trombofilia a los familiares de primer grado de personas con antecedentes de trombosis venosa profunda o embolia pulmonar y trombofilia.
Embarazo, anticoncepción y tratamiento de reemplazo hormonal
La anticoncepción y el embarazo en familiares mujeres en edad fértil y el reemplazo hormonal en mujeres mayores se asocian con problemas específicos Según una revisión reciente, los anticonceptivos orales combinados aumentan el riesgo anual de TEV de 2 en 10000 a aproximadamente 5-10 en 10000 mujeres. Los riesgos parecen ser mayores en mujeres con trombofilias hereditarias de familias proclives a la trombosis. La revisión de los anticonceptivos hormonales combinados por la European Medicines Agency indica que no se deben recetar anticonceptivos orales combinados a una mujer con predisposición para un trastorno de la coagulación y que la conveniencia de éstos se debe analizar si la mujer tiene un familiar cercano que haya sufrido un episodio tromboembólico antes de los 50 años. Puesto que las mujeres con antecedentes de TEV en un familiar de primer grado antes de los 50 años deben considerar otra forma de anticoncepción, no son necesarias las pruebas para trombofilia.
Las mujeres con antecedentes de TEV no provocada en un familiar de primer grado no deben emplear tratamiento de reemplazo hormonal, pero si éste se considera esencial, se les puede ofrecer tratamiento transdérmico, que parece no tener riesgo significativo de TEV.
Se debe evaluar el riesgo de trombosis venosa asociada con el embarazo en las embarazadas o las que planean un embarazo y tienen un familiar de primer grado con TEV no provocada o asociada con estrógenos. Esto debe incluir una pesquisa para trombofilia, ya que las recomendaciones del Royal College of Obstetricians and Gynaecologists sugieren tromboprofilaxis prenatal si estas mujeres tienen deficiencia de antitrombina u homocigosidad o doble heterocigosidad para trombofilia hereditaria, aunque las recomendaciones de los EEUU aconsejan tromboprofilaxis prenatal sólo ante la homocigosidad para el factor V de Leiden o la mutación del gen de protrombina. Se debe considerar la tromboprofilaxis para todas las mujeres con antecedentes familiares de TEV y trombofilia ya conocida, especialmente ante otros factores de riesgo.
Púrpura fulminante
Una situación infrecuente donde las pruebas para trombofilia son importantes es en neonatos con purpura fulminante, que se puede producir por deficiencias homocigóticas de proteína C o proteína S. La confirmación del diagnóstico permite administrar tratamiento de reemplazo precoz, así como considerar el tratamiento prolongado.
Enfermedad arterial
Se mencionaron asociaciones estadísticamente significativas, pero no importantes desde el punto de vista clínico, entre enfermedad coronaria y factor V de Leiden y la mutación del gen de la protrombina. La importancia de las trombofilias hereditarias es pequeña en relación con los factores de riesgo tradicionales para enfermedad arterial.
Las recomendaciones británicas indican que los estudios para trombofilia hereditaria no están indicados en la enfermedad arterial.
Morbilidad en el embarazo
Aunque el síndrome antifosfolipídico (una trombofilia adquirida) se asocia con complicaciones del embarazo, tales como aborto espontáneo y preclampsia, los estudios de casos y controles sobre el tema tuvieron resultados variables. Las recomendaciones de los EEUU no aconsejan el tratamiento antitrombótico en mujeres con trombofilia hereditaria y antecedentes de complicaciones del embarazo. Sin embargo, un estudio de observación reciente indicó que la heparina de bajo peso molecular se asoció con disminución del riesgo de muerte fetal y de preclampsia grave de inicio precoz en los embarazos ulteriores de mujeres que habían sufrido una única muerte fetal por lo demás inexplicable a las 10 o más semanas de embarazo y padecían trombofilia hereditaria. En un metanálisis reciente de estudios aleatorizados de heparina de bajo peso molecular en mujeres con complicaciones en embarazos anteriores causadas por la placenta, el 25% de las cuales tenían trombofilia, se sugiere que el tratamiento puede reducir los resultados adversos recurrentes, como la preclampsia, el aborto espontáneo en los últimos meses de embarazo y el nacimiento de neonatos de bajo peso para la edad gestacional. Aunque estos informes pueden renovar el interés en la heparina de bajo peso molecular para prevenir las complicaciones recurrentes del embarazo, aún no se conoce la importancia de las pruebas para la trombofilia hereditaria y es necesario continuar las investigaciones antes de considerar esta intervención.
♦ Traducción y comentario objetivo: Dr. Ricardo Ferreira.
