La enfermedad de Fabry (FD) es una enfermedad de depósito lisosomal rara, ligada al X causada por mutaciones en el gen de la a-galactosidasa (GLA) ocasionando una deficiencia de esa enzima. La FD puede presentar consecuencias serias, con daño y falla de órganos, y por lo tanto es esencial la intervención temprana. Es importante que los médicos consideren el diagnóstico en mujeres, las que generalmente tienen manifestaciones más sutiles que los hombres y que sean capaces de reconocer otras características de FD como la apariencia facial típica, pérdida de audición neurosensorial, fenómeno de Raynaud (RP) e hipohidrosis. Se reporta el caso de una mujer con historia familiar de FD, que presentaba la facie característica de la enfermedad, como su hijo.
Reporte del caso:
Se presenta una mujer de 55 años (Fig 1 a y b) con empeoramiento de los síntomas de RP en las manos. Presentaba hipohidrosis, intolerancia al calor, fibrilación auricular e hipotiroidismo. Manifestaba dolor intermitente y parestesia en pies y edema de tobillos, disnea al esfuerzo y diarrea.

Figura 1 (a,b) Características faciales de la enfermedad de Fabry en la paciente.
Refirió que los cuatro hermanos tenían problemas cardíacos. Sus dos hijos, de 34 y 36 años de edad, manifestaban hipohidrosis, edema de tobillos y enfermedad renal crónica estadío 2. Ambos presentaban similares características a la madre (Fig 2). Un hijo con numerosos angioqueratomas más de 100 en el área “calzón de baño”, en miembros, incluyendo palmas, plantas y dedos de pies. El otro presentaba menos angioqueratomas (50-100) con una distribución similar.
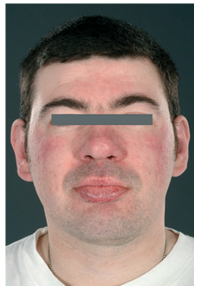
Figura 2: Características faciales de la enfermedad de Fabry en el hijo de la paciente.
Al examen físico se observaba , arcos supraorbitarios prominentes, cejas tupidas, puente nasal ancho y prognatismo, 50 a 100 angiomas rojos y otros color cereza diseminados en tronco y miembros proximales. No se observaban angioqueratomas clásicos.
Los análisis de laboratorio indicaron falla cardíaca y dilatación leve biauricular, deterioro de la función renal, pérdida de audición neurosensorial unilateral leve e hipercolesterolemia.
El nivel de a-galactosidasa plasmático estaba en el nivel inferior del rango normal (4.3 nmol/h/mL; rango normal 4-21.9 nmol/h/mL) y el nivel de a-galactosidasa leucocitaria era normal (69 nmol/h/mg/proteína; normal 33-134 nmol/h/mg/proteína). Este es el caso en aproximadamente el 30% de las mujeres afectadas, y el análisis de ADN es mandatario para excluir el diagnóstico.
El análisis de genes reveló delección en el exon 1 del gen de GLA, que codifica la a-galactosidasa, confirmando el diagnóstico de FD. Se encontró la misma delección en los dos hijos.
Se inició tratamiento con perindopril, rosuvastatina, aspirina y terapia de reemplazo hormonal con agalsidasa alfa. Los síntomas de RP se controlaron con 60 mg de diltiazem diarios.
En su forma clásica, la prevalencia de de FD es entre 1 en 40.000 y 1 en 476000. Los estudios de screening neonatal recientes sugieren que en el inicio tardío, las formas leves de la enfermedad pueden ser prevalentes 1 en 1500 a 1 en 3700 hombres. La reducción o ausencia de la actividad de la enzima ocasiona la acumulación de globotriaosilceramida (Gb)3 en los lisosomas del cuerpo, dando lugar a una amplia variedad de síntomas clínicos progresivos
Los hallazgos tempranos incluyen acroparestesias, problemas gastrointestinales, hipohidrosis y angioqueratomas. Los signos y síntomas tardíos de falla de órgano pueden manifestarse como proteinuria y glomeruloesclerosis, hipertrofia ventricular izquierda, arritmias cardíacas y accidente cerebrovascular.
Este caso ilustra una mujer que puede tener características severas de FD, pero también que dos tercios de las mujeres con FD no tienen angioqueratomas.
También destaca una de las características menos reconocidas de la enfermedad: “apariencia facial pseudoacromegálica”, RP e hipohidrosis.
La facie de FD parece ser más común en hombres con angioqueratomas diseminados y enfermedad clásica. Dos estudios independientes han identificado que comparado con los controles, los pacientes masculinos con FD tienen amplitud periorbitaria, arcos supraorbitarios prominentes, incremento de la amplitud bitemporal, cejas tupidas, ptosis, base nasal ancha, labios gruesos y prognatismo.
Las características en mujeres son similares pero más sutiles, y pueden ser subdiagnosticadas.
La intolerancia al frío y la manifestación de dolor en los miembros con el frío es un síntoma común en pacientes con FD. Más recientemente, se documentó RP en el 8% de 710 mujeres y en el 11% de 644 hombres con Fabry. En un subgrupo de 98 pacientes (58 mujeres), el 34% de las mujeres y el 25% de los hombres describieron dedos fríos, azules, dolorosos en verano e invierno. En la mitad de los casos, los cambios de color bifásicos o trifásicos con dolor estaban presentes al aumentar la temperatura.
La sudoración disminuída y la intolerancia al calor son características clásicas de la FD. Ocurre en el 53% de los varones y en el 28% de las mujeres, probablemente como consecuencia de una neuropatía autonómica.
También pueden presentar hiperhidrosis, y es más común en mujeres. El tinitus y pérdida de audición para tonos altos puede ser de inicio repentino, y es una característica bien reconocida de la enfermedad.
Existe un tratamiento efectivo para FD en la forma de terapia de reemplazo enzimático.
Es importante conocer todas las características de la enfermedad, hasta las menos frecuentes, para realizar un diagnóstico más temprano y un tratamiento precoz, que reduce la morbilidad y mortalidad.
La presencia de las características faciales mencionadas, particularmente si se asocian con síntomas cardíacos, renales u otros como RP, sordera o anormalidades del sudor, deben incentivar al estudio de una probable FD en pacientes masculinos y femeninos, aún en ausencia de angioqueratomas típicos.
¿Qué aporta éste artículo a la práctica dermatológica?.
Aunque la enfermedad de Fabry (FD) es una enfermedad de depósito lisosomal ligada al X, existe una alta prevalencia de mujeres que muestran síntomas y presentan incremento de la mortalidad asociada con la enfermedad. La FD generalmente progresa lentamente, y las causas de muerte pueden ser accidente cerebrovascular, enfermedad cardiovascular o falla renal. El diagnóstico puede retrasarse en pacientes femeninas que presentan generalmente características sutiles. El fenotipo cutáneo clásico de angioqueratoma corporal difuso es menos común en pacientes femeninas.
Se reporta el caso de una mujer con historia familiar de FD, que mostró algunas características menos reconocidas de FD, incluyendo la apariencia típica de pseudo acromegalia facial.
Presentaba delección en el exon 1 del gen a-galactosidasa (GLA), confirmando el diagnóstico de FD. Como en el 30% de las mujeres con FD, los niveles de a-galactosidasa plasmáticos y leucocitarios se encontraban en el límite inferior del rango normal. A la presentación, tenía síntomas y signos de daño de órgano.
Las mujeres heterocigotas pueden presentar síntomas de FD y tener un incremento de la mortalidad asociada a la enfermedad.
Los médicos deben tener en cuenta la apariencia facial típica de FD. El diagnóstico y tratamiento temprano de FD puede reducir la morbilidad y mortalidad de la enfermedad.
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello
