Introducción
El síndrome de Guillain-Barré se caracteriza por debilidad aguda progresiva, arreflexia, y máxima discapacidad motora que ocurre dentro de las 4 semanas posteriores al inicio del cuadro. Es una neuropatía periférica aguda, post-infecciosa, mediada por mecanismos inmunes, con curso clínico y resultados muy variables. Se han descripto varios subtipos clínicos desde la descripción original del síndrome. Las diferentes variantes se caracterizan por el compromiso local o regional de los nervios periféricos y autonómicos. Debido a que no existe un solo marcador clínico o serológico para el síndrome de Guillain-Barré, el diagnóstico se basa en los hallazgos clínicos, de laboratorio, y neurofisiológicos, junto con la exclusión de otras condiciones que se asemejan con la enfermedad.
El síndrome de Guillain-Barré es reconocido como un trastorno heterogéneo con diversas manifestaciones clínicas. Recientes hallazgos neurofisiológicos y patológicos llevaron a su reclasificación, ampliando el espectro clínico y patológico desde la clásica polineuropatía desmielinizante inflamatoria aguda y sus variantes axonales hasta variantes clínicas severas (por ejemplo, el síndrome de Miller Fisher, la encefalitis del tronco cerebral de Bickerstaff, la variante faringo-cérvico-braquial, la polineuritis craneal, y la neuropatía sensorial aguda). El no reconocer estas variantes del síndrome de Guillain-Barré puede dar lugar a diagnósticos erróneos, tratamientos inadecuados, y a una morbilidad significativa.
El presente estudio tuvo como objetivo describir las manifestaciones clínicas, los hallazgos de laboratorio y de imágenes, el tratamiento y el pronóstico en niños que manifiestan variantes clínicas del síndrome de Guillain-Barré.
Métodos
El diagnóstico de síndrome de Guillain-Barré agudo se basó principalmente en la sintomatología y en los datos auxiliares de apoyo, incluyendo los resultados del líquido cefalorraquídeo, las evaluaciones electrofisiológicas o de imágenes. Se incluyeron niños previamente sanos menores de 18 años de edad con diagnóstico al alta de síndrome de Guillain-Barré, pero no aquellos con polineuropatía desmielinizante inflamatoria crónica o con condiciones conocidas que causan polineuropatía aguda, o con una enfermedad subyacente anterior como por ejemplo enfermedad autoinmune, injuria neurológica previa, o un trastorno neurológico progresivo.
Se revisaron retrospectivamente los estudios de electrodiagnóstico y las historias clínicas de los pacientes con síndrome de Guillain-Barré admitidos en el Hospital de Niños Chang Gung entre enero de 2000 y junio de 2010. Se enrolaron 43 niños después de haber sido examinados por un neurólogo. La información recopilada incluyó edad al momento de la presentación, sexo, subtipo de clasificación, infección previa (tracto respiratorio o gastrointestinal), tiempo desde el inicio de los signos hasta su nadir, afectación de pares craneales (por ejemplo, alteración del reflejo de deglución, disfagia o disartria), disfunción autonómica (por ejemplo, fluctuaciones excepcionales de la presión arterial o arritmia cardiaca inexplicable), gravedad clínica máxima, neuroimágenes, tratamiento específico (es decir, corticoides intravenosos, inmunoglobulina, o plasmaféresis), duración de la estadía hospitalaria, y pronóstico.
Las variantes del síndrome de Guillain-Barré se dividieron inicialmente en dos grupos de acuerdo a la presentación clínica, ya sea como forma clásica ascendente o como formas de presentación atípica. La forma ascendente típica del síndrome de Guillain-Barré se caracterizó por paresia fláccida ascendente, progresiva y simétrica, con arreflexia. Se presenta un diagrama de flujo diagnóstico en la Figura 1. El grupo de presentación atípica del síndrome de Guillain-Barré se caracterizó por compromiso localizado o regional de los axones motores y sensoriales de los nervios periféricos y del sistema nervioso autónomo. En el grupo ascendente típico, se clasificaron dos subtipos de acuerdo con los estudios electrodiagnósticos: (1) las formas desmielinizantes (por ejemplo, polirradiculoneuropatía desmielinizante inflamatoria aguda) y (2) las formas axonales, que incluyeron la neuropatía axonal aguda motora-sensorial y la neuropatía axonal motora aguda. En el grupo de presentación atípica, se clasificaron dos subtipos según la presentación atípica y los hallazgos neurofisiológicos: (1) compromiso importante del nervio craneal, incluyendo el síndrome de Miller Fisher, la encefalitis del tronco cerebral de Bickerstaff, la variante faringo-cérvico-braquial, y la polineuritis craneal; y (2) otros, incluyendo la disautonomía general aguda y la neuropatía sensorial aguda.
Para determinar si las manifestaciones clínicas y los resultados de las variantes clínicas del síndrome de Guillain-Barré diferían del síndrome típico, la polirradiculoneuropatía desmielinizante inflamatoria aguda se definió como síndrome de Guillain-Barré típico, y las otras formas de presentación (incluyendo las formas axonales y las presentaciones atípicas) se definieron como variantes clínicas. El pico de gravedad clínica se cuantificó por el grado de debilidad máxima del miembro superior, de acuerdo con una escala ordinal, es decir: 0, normal; 1, débil, pero capaz de levantar los brazos fuera de la cama; 2, movimiento de parpadeo; 3, sin movimiento. Se evaluaron los resultados 1 año después del inicio de la enfermedad. Los resultados funcionales se clasificaron de acuerdo a la escala de Hughes y col. adaptada por los autores, es decir: 0, sanos; 1, signos menores, capaz de correr; 2, capaz de caminar más de 5 metros sin asistencia, pero incapaz de correr; 3, capaz de caminar más de 5 m con asistencia; 4, permanencia en cama o silla; 5, requerimiento de ventilación asistida al menos parte del día; y 6, fallecimiento. Se definió como resultado favorable a la independencia funcional y a la capacidad para deambular de forma independiente (puntuación de Hughes 0-2). El estudio fue aprobado por la Junta de Revisión Institucional del Hospital de los autores.
Análisis estadístico
Las características clínicas, bioquímicas, electrofisiológicas y los resultados de las variantes clínicas del síndrome de Guillain-Barré se compararon con aquellas del síndrome de Guillain-Barré típico. Las características clínicas obtenidas al momento del ingreso se analizaron para buscar las posibles diferencias entre los dos grupos. Estas características incluyeron edad, sexo, infección previa, subtipo de síndrome de Guillain- Barré, tiempo desde el inicio de los signos hasta la admisión hospitalaria, afectación de los pares craneales, disautonomía, máxima gravedad clínica, neuroimágenes, y tratamiento específico. También se examinaron las asociaciones entre cada una de estas variables y los resultados.
El análisis estadístico se realizó mediante el programa de software estadístico SPSS versión 12.0 (SPSS Inc., Chicago, IL). Los factores predictivos univariados de los resultados (buenos o malos) se evaluaron mediante la prueba U de Mann-Whitney para las variables continuas y la prueba exacta de Fisher para las variables categóricas. Se consideró estadísticamente significativa una P < 0,05. Todas las pruebas estadísticas fueron de dos colas.
Resultados
Datos demográficos
Se enrolaron 43 pacientes con síndrome de Guillain-Barré, basado en datos clínicos y electrofisiológicos, con edades comprendidas entre 1 año y 8 meses a 18 años (media ± desvío estándar, 7,88 ± 5,31 años). Se incluyeron 13 (30,2%) niñas y 30 (69,8%) niños. Diecinueve (44,2%) eran menores de 4 años, 11 (25,6%) tenían entre 5 y 8 años, uno (2,3%) estaba en el grupo de edad de 9 a 12 años, y 12 (27,9%) eran mayores de 13 años. La mayor incidencia de síndrome de Guillain-Barré se produjo en el grupo de menores de 4 años. Aproximadamente dos terceras partes de los pacientes eran menores de 8 años.
Clasificación
En el grupo clásico ascendente, 29 pacientes (67,4%) manifestaron una polirradiculoneuropatía desmielinizante inflamatoria aguda, 2 (4,7%) presentaron neuropatía axonal motora aguda, y 1 (2,3%) neuropatía axonal motora sensorial aguda.
En el grupo de presentación atípica, 11 pacientes (25,6%) manifestaron una participación importante de los pares craneales, incluyendo 4 (9,3%) con síndrome de Miller Fisher, 3 (7%) con encefalitis del tronco cerebral de Bickerstaff, 2 (4,7%) con la variante faringo-cérvico-braquial, y 2 (4,7%) con polineuritis craneal. No hubo diferencias significativas evidentes entre la edad y la clasificación.
Manifestaciones clínicas
El signo inicial más frecuente fue la debilidad de las extremidades, documentado en 100 (100%) pacientes, seguido por dolor o malestar muscular (n=19; 44,2%) y parestesia (n=18; 41,9%). El tiempo medio desde el comienzo de la enfermedad hasta su nadir en los 43 pacientes fue de 5,69 días (rango, 1-20 días), dividido en 6,79 días (rango, 1-20 días) en el síndrome de Guillain-Barré típico y 3,50 días (rango, 1-10 días) en el grupo de variantes clínicas. El tiempo desde el inicio de la enfermedad hasta el punto de menor intensidad fue menor en el grupo de variantes clínicas que en el grupo de la forma clásica ascendente, pero no hubo diferencias significativas evidentes entre estos dos grupos (P=0,014). La puntuación media de discapacidad en el nadir ± desvío estándar fue de 0,83 ± 0,80 en el síndrome de Guillain-Barré típico, y de 1,93 ± 0,92 en el grupo de variantes clínicas de acuerdo al pico de gravedad clínica.
Los autores puntuaron 28 pacientes con grado 3 o más durante su hospitalización en base a la escala de clasificación funcional de Hughes y colaboradores. La puntuación en el síndrome de Guillain-Barré típico fue de 2,76 ± 0,91, y la puntuación en el grupo de variantes clínicas fue de 4 ± 0,96, según la escala de Hughes y colaboradores. Se evidenciaron diferencias significativas entre estos dos grupos en la puntuación media de discapacidad y en la escala de Hughes en el nadir (P=0,004 y 0,004, respectivamente).
Otros signos incluyeron afectación de pares craneales (n=17; 39,5%), disfunción autonómica (n=13; 30,2%), incontinencia urinaria (n=11; 25,6%) y dolor de cabeza/mareos (n=9; 20,9%). Los niños del grupo de variantes clínicas tuvieron más probabilidades de manifestar afectación de nervios craneales e incontinencia urinaria que las personas con síndrome de Guillain-Barré típico (P ≤ 0,001, y < 0,001, respectivamente). Se evidenciaron signos respiratorios, incluyendo opresión torácica o disnea, en 10 pacientes (23,3%). Siete (16,3%) requirieron apoyo ventilatorio durante la hospitalización. En el grupo de variantes clínicas, el número de pacientes con síntomas respiratorios (7 de 14 niños) y de pacientes con apoyo ventilatorio (5 de 14 niños) fue significativamente mayor que en el síndrome de Guillain-Barré típico (3 de 29 niños y 2 de 29 niños, P=0,007 y 0,028, respectivamente). Ningún paciente falleció durante la hospitalización.
Preponderancia estacional e infección precedente
Nueve pacientes desarrollaron Guillain-Barré durante la primavera (marzo-mayo), 12 durante el verano (junio-agosto), 10 durante el otoño (septiembre-noviembre), y 12 durante el invierno (diciembre-febrero). El grupo de variantes clínicas en su mayoría desarrolló la enfermedad en verano e invierno, y fue evidente la diferencia significativa entre estos dos grupos. Cuatro pacientes (9,3%) no tuvieron ningún evento desencadenante conocido en las 4 semanas previas a la aparición del síndrome de Guillain-Barré. De los 39 pacientes (90,7%) con factores desencadenantes, 28 pacientes presentaron estos eventos en las 2 semanas previas a la aparición del síndrome de Guillain-Barré, y 11 pacientes tuvieron estos eventos entre las 2-4 semanas previas a la aparición del mismo. Las infecciones del tracto respiratorio superior (86%, 37/43) comprendieron la mayor parte de los desencadenantes más frecuentes, seguidas por la gastroenteritis aguda (4,7%, 2/43). Además, ni los antecedentes familiares de esta enfermedad, ni la enfermedad en otros miembros de la familia antes o durante la aparición de los signos sugirieron la posibilidad de un virus neuropático u otra infección. Ninguno había recibido vacunas previamente.
Entre los 37 pacientes con infección de las vías respiratorias superiores en el grupo con síndrome de Guillain-Barré, 12 estaban infectados con Mycoplasma pneumoniae (32,4%), 5 con virus Epstein-Barr (13,5%), 3 con virus del herpes humano-6 (8,1%), 2 con influenza A (5,4%), y otro con el virus Varicela zoster (2,7%). No se identificó ningún agente patógeno en los otros 14 pacientes.
Líquido cefalorraquídeo y neuroimágenes
Se realizó estudio del líquido cefalorraquídeo en 34 pacientes. El nivel de proteínas totales del líquido cefalorraquídeo osciló entre 15,5 y 300 mg/dl, con un recuento de leucocitos de 0-28 células/mm3. La concentración media de proteínas del líquido cefalorraquídeo en los 34 pacientes fue de 107,32 mg/dl (79,1%, 34/43). Se detectó un recuento medio de 4,72 células/mm3 en el líquido cefalorraquídeo de 25 pacientes, y todos presentaron disociación albúmino-citológica (es decir, una concentración elevada de proteínas, sin pleocitosis). Los resultados del líquido cefalorraquídeo no fueron diferentes entre el grupo de síndrome de Guillain-Barré típico y el grupo de variantes clínicas.
Se realizó resonancia magnética espinal en 21 pacientes. En el grupo de síndrome de Guillain-Barré típico, la resonancia magnética espinal con gadolinio (n = 9) reveló un realce de la raíz radicular del nervio espinal en dos pacientes y de la cola de caballo en otros dos, y se obtuvieron resultados negativos en los cinco restantes. En el grupo de variantes clínicas, la resonancia magnética espinal indicó hiperintensidad en el tronco cerebral en T2 (n=3), y realce de la raíz radicular de la cola de caballo en la resonancia magnética de la columna vertebral con gadolinio (n=2).
Tratamiento
Los diversos regímenes terapéuticos utilizados en este estudio incluyeron plasmaféresis terapéutica en dos pacientes (4,7%), inmunoglobulina intravenosa sola en ocho (18,6%), corticoides solos en 22 (51,2%), corticoides y plasmaféresis o inmunoglobulina intravenosa combinados en ocho (18,6%), y tratamiento de apoyo en tres (7%). En el grupo de variantes clínicas, se utilizó corticoides solos en siete pacientes, inmunoglobulina intravenosa sola en tres, corticoides y plasmaféresis o inmunoglobulina intravenosa combinados en tres, y tratamiento de apoyo en uno.
Resultados al año de seguimiento
Se analizaron los aspectos funcionales de los pacientes en el seguimiento después de 1 año o más de la aparición del cuadro. El estudio de seguimiento reveló que 37 pacientes se recuperaron satisfactoriamente y 6 permanecieron con paresia residual y marcha asistida. No se registró mortalidad durante los ingresos hospitalarios. El grupo de variantes clínicas, después de la fase aguda, abarcó a la mayoría de los niños que requirieron rehabilitación extendida. Ocho pacientes se recuperaron, y seis quedaron con parálisis residual y marcha asistida. De los seis con paresia residual, dos manifestaron la variante faringo-cérvico-braquial, dos manifestaron encefalitis del tronco cerebral de Bickerstaff, uno manifestó neuropatía axonal aguda motora-sensorial, y uno manifestó neuropatía motora axonal aguda. En el síndrome de Guillain-Barré típico se recuperaron todos los pacientes.
Los pacientes se dividieron en dos grupos en base al estado funcional (favorable, puntaje de Hughes 0-2; pobre, puntaje de Hughes 3-5), y se compararon de acuerdo a su condición después de 1 año. La media de la puntuación de discapacidad en el grupo de variantes clínicas fue significativamente peor que en el grupo de síndrome de Guillain-Barré típico (P < 0,001). Los malos resultados fueron probablemente atribuibles a los signos respiratorios (odds ratio, 32,00; intervalo de confianza 95%: 3,07 a 333,79; P=0,001), la necesidad de asistencia respiratoria (odds ratio, 8,25; intervalo de confianza 95%: 1,22 a 55,56; P=0,045), la incontinencia urinaria (odds ratio, 2,20; intervalo de confianza 95%: 1,15 a 4,20; P <0,001), y a la etiología (grupo de variantes clínicas; odds ratio, 1,75; intervalo de confianza 95%: 1,11 a 2,75; P <0,001).
Los autores también dividieron a los pacientes en un grupo tratado sólo con esteroides y en un grupo tratado sólo con inmunoglobulina intravenosa/plasmaféresis, y compararon los resultados al año de seguimiento. Los resultados no fueron estadísticamente diferentes entre estos dos grupos.
Discusión
El síndrome de Guillain-Barré es una polineuropatía inflamatoria aguda. Clínicamente, la debilidad motora y la pérdida sensorial comienzan en las extremidades inferiores, y de manera simétrica y progresiva ascienden a las extremidades superiores. El diagnóstico es relativamente fácil en los pacientes con presentaciones clínicas y hallazgos neurofisiológicos típicos. Sin embargo, como cada vez más la investigación clínica se centra en esta enfermedad, se tiene cada vez más claro que el síndrome de Guillain-Barré no es una entidad única, sino más bien un grupo clínica y patológicamente heterogéneo de condiciones neuropáticas. Un número de variantes del síndrome de Guillain-Barré se caracteriza por afectación localizada o regional de los nervios periféricos y autónomos. El reconocimiento de los casos atípicos es importante porque permite el monitoreo anticipado de complicaciones, e identifica opciones terapéuticas para los niños afectados.
Desde un punto de vista clínico, los criterios diagnósticos del síndrome de Guillain-Barré no abarcan la totalidad del espectro clínico de este problema, y el diagnóstico se complica aún más cuando el cuadro clínico no es clásico. Por otro lado, una presentación que en términos generales se ajusta ampliamente a los límites clínicos del síndrome de Guillain-Barré puede causar una tendencia a pasar por alto otros posibles diagnósticos, algunos de los cuales pueden provocar la muerte si no se reconocen en forma suficientemente precoz. En 1993, se ampliaron los criterios diagnósticos del síndrome de Guillain-Barré bajo los auspicios de la Organización Mundial de la Salud. Estos criterios ahora se basan exclusivamente en factores clínicos. Como consecuencia, los subgrupos se proponen de una manera descriptiva. Estos nuevos criterios no resuelven la cuestión de cómo juzgar todas las diferentes características que pueden ocurrir en pacientes individuales. Se ha diseñado un esquema de subclasificación para cubrir las variantes clínicas de una manera sistemática.
En el presente estudio, se desarrolló un diagrama de flujo diagnóstico para el síndrome de Guillain- Barré teniendo en cuenta las clasificaciones ya sugeridas en la literatura, y garantizando que la clasificación propuesta sea capaz de cubrir todas las variantes del síndrome. En el primer paso de este diagrama de flujo diagnóstico, los pacientes se clasifican en dos grupos según su presentación clínica, ya sea como la típica forma ascendente o como presentación atípica. En segundo lugar, para el grupo de forma ascendente típica, los subtipos se clasifican en base al estudio electrodiagnóstico en la forma desmielinizante y en la axonal. En el grupo de presentación atípica, los subtipos se clasifican de acuerdo a la presentación clínica atípica, y los hallazgos neurofisiológicos se clasifican según la afectación de los pares craneales y de otros. Las diferencias fundamentales entre estos subtipos se observan principalmente en la presentación clínica y en el estudio electrodiagnóstico.
En las primeras etapas, el reconocimiento de las variantes clínicas del síndrome de Guillain- Barré puede ser muy difícil. Las principales características del síndrome de Guillain-Barré incluyen la debilidad bilateral rápidamente progresiva y simétrica de las extremidades, con o sin afectación de los músculos respiratorios o de los pares craneales. En los casos típicos, los síntomas asociados son dolor, entumecimiento y parestesia. Sin embargo, en las variantes clínicas, algunas características traerían dudas acerca del diagnóstico de Guillain-Barré. Las presentaciones atípicas incluyen debilidad asimétrica, debilidad que involucra inicialmente sólo los brazos, deterioro rápidamente progresivo de la función pulmonar con una relativa preservación de la fuerza muscular en las extremidades, y disfunción autonómica. De acuerdo con la experiencia clínica del presente estudio, los niños con variantes clínicas son más propensos que los niños con el típico síndrome de Guillain-Barré a manifestar una rápida progresión desde el inicio de la enfermedad hasta el nadir, con aumento de la severidad en las puntuaciones medias de discapacidad y en la escala de Hughes en el nadir, afectación de los pares craneales, e incontinencia urinaria (P=0,014, P=0,004, P=0,004, P<0,001, y P<0,001, respectivamente).
Los niños con síndrome de Guillain-Barré presentan un riesgo bajo de complicaciones respiratorias, tales como insuficiencia respiratoria con requerimiento de ventilación asistida durante la hospitalización. Sólo el 13% de los pacientes pediátricos necesitaron asistencia respiratoria durante la hospitalización. En el presente estudio, siete pacientes (16,3%) recibieron apoyo de un respirador. En el grupo de variantes clínicas, el número de pacientes con signos de insuficiencia respiratoria (siete de 14 niños) y asistencia respiratoria (cinco de 14 niños) fue significativamente mayor que en aquellos con síndrome de Guillain-Barré típico (tres de 29 niños y dos de 29 niños, P=0,007 y 0,028, respectivamente). Con la asistencia respiratoria, estos niños demostraron una buena recuperación de las graves complicaciones respiratorias de la fase aguda, y todos fueron extubados. Así, al inicio de la enfermedad, el rápido progreso desde su aparición al nadir, el aumento de la gravedad de las medias de las puntuaciones de discapacidad y de la escala de Hughes en el nadir, la afectación de los pares craneales, la incontinencia urinaria, los síntomas respiratorios, y la necesidad de soporte ventilatorio pueden ser útiles en el reconocimiento temprano de las variantes de las formas clínicas del síndrome de Guillain-Barré.
Los corticoides no se recomiendan para el tratamiento del síndrome de Guillain-Barré, y dicho tratamiento ha sido rigurosamente estudiado en adultos. Sin embargo, la inmunoterapia en niños con síndrome de Guillain-Barré no se estudió en profundidad con estudios aleatorizados y bien controlados. La plasmaféresis y la inmunoglobulina intravenosa constituyen en la actualidad los tratamientos recomendados en este grupo de edad.
En conclusión, durante la etapa temprana del síndrome de Guillain-Barré, la gravedad en aumento en las puntuaciones medias de discapacidad y en la escala de Hughes en el nadir, la afectación de los pares craneales, la incontinencia urinaria, los síntomas respiratorios, y la necesidad de asistencia respiratoria se asocian con peor pronóstico. El reconocimiento de las presentaciones atípicas del síndrome de Guillain-Barré es importante para determinar los tratamientos adecuados y mejorar el pronóstico de esta enfermedad.
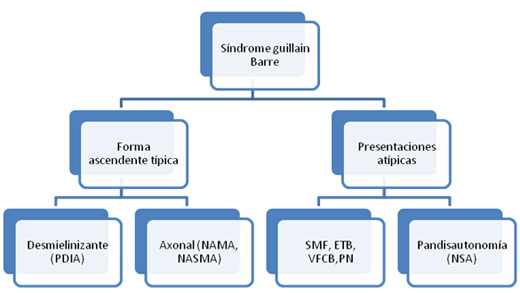
Figura 1. Diagrama de flujo diagnóstico del síndrome de Guillain Barré y sus variantes. El grupo típico incluye (1) Poliradiculoneuropatía desmielinizante inflamatoria aguda (PDIA), y (2) Neuropatía axonal sensitivo motora aguda (NASMA) y neuropatía axonal motora aguda (NAMA). El grupo atípico incluye (1) Síndrome de Miller Fisher (SMF), encefalitis del tronco cerebral de Bickerstaff (ETB), variante faringo cérvico braquial (VFCB), y polineuritis craneal (PN), y (2) Pandisautonomía aguda y neuropatía sensorial aguda (NSA).
Comentario: El presente estudio describe las variantes clínicas del síndrome de Guillain Barré, tanto en sus formas típicas como atípicas, haciendo mayor hincapié en la importancia del reconocimiento de las formas menos habituales para lograr un correcto diagnóstico diferencial. Se remarca la importancia del diagnóstico precoz para la instauración del tratamiento adecuado y mejorar el pronóstico a largo plazo.
♦ Resumen y comentario objetivo: Dra. Alejandra Coarasa