Introducción
La insuficiencia suprarrenal (IS) es un trastorno que puede causar la muerte, provocado por la insuficiencia suprarrenal primaria (ISP) o la enfermedad suprarrenal secundaria a la alteración del eje hipotálamo-hipofisario. Es la manifestación clínica de la deficiente producción o acción de los glucocorticoides, con o sin deficiencia de mineralocorticoides y andrógenos suprarrenales.
Manifestaciones clínicas

Independientemente de la causa, la IS ha sido fatal hasta el año1949, cuando fue sintetizado por primera vez el cortisol y se pudo comenzar a hacer el tratamiento de reemplazo de los glucocorticoides. Sin embargo, a pesar de este avance, el diagnóstico y el tratamiento de los pacientes sigue siendo problemático.
Epidemiología
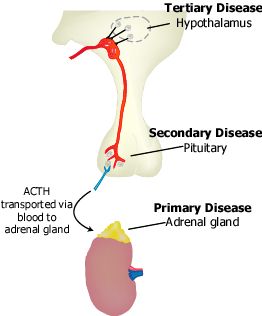 Según el mecanismo subyacente, la IS se clasifica en primaria, secundaria o terciaria.
Según el mecanismo subyacente, la IS se clasifica en primaria, secundaria o terciaria.
- La insuficiencia suprarrenal primaria (ISP) resulta de la enfermedad intrínseca de la corteza suprarrenal.
- La IS central, es el conjunto de las insuficiencias secundaria y terciaria, está causada por el deterioro de la producción o de la acción de la corticotrofina (ACTH).
- La IS secundaria está causada por la enfermedad de la hipófisis que obstaculiza la liberación de ACTH o por la incapacidad de las glándulas suprarrenales para responder a esa hormona.
- La IS terciaria resulta de la alteración hipotalámica de la síntesis o la acción de la hormona liberadora de ACTH, la vasopresina arginina o ambas, la que a su vez inhibe la secreción de ACTH.
Hacia fines del siglo XX, la prevalencia de la ISP crónica en Europa se duplicó y actualmente se estima en 4,4-4,6 casos nuevos por millón por año. Durante la primera mitad del siglo XX, la causa más común de ISP fue la tuberculosis pero en la actualidad son más comunes las enfermedades autoinmunes.
El aumento de la frecuencia de ISP en las últimas décadas, asociado la declinación de la prevalencia de la tuberculosis indica que hay más casos de IS autoinmune. En una serie de 615 pacientes con enfermedad de Addison estudiados entre 1969 y 2009, el 82% correspondió a la forma autoinmune; el 9% a casos relacionados con la tuberculosis y alrededor del 8% a otras causas. La ISP ocurre con más frecuencia en las mujeres y puede presentarse a cualquier edad, aunque predomina entre los 30 y los 50 años.
La frecuencia de las diversas formas de ISP en los niños difiere sustancialmente de la población adulta. En los niños, son más comunes las formas genéticas. En una serie de casos de 103 niños con enfermedad de Addison, observados durante 20 años (1981–2001), la causa más frecuente fue la hiperplasia suprarrenal congénita (72%); otras alteraciones genéticas eran responsables de otro 6% mientras que la enfermedad autoinmune fue diagnosticada solamente en el 13% de los casos.
La IS secundaria es más común que la ISP y su prevalencia ha sido estimada en 150–280 por millón de personas y afecta con más frecuencia a las mujeres.
La edad pico en el momento del diagnóstico es la sexta década de la vida. Una revisión sistemática y metaanálisis de prevalencias de hipopituitarismo en pacientes adultos que recibieron irradiación craneana por tumores no hipofisarios mostró una prevalencia puntual de cualquier grado de hipopituitarismo de 0,66 y una prevalencia de deficiencia de ACTH de 0,.22.
La causa más común de IS terciaria es la administración prolongada de glucocorticoides exógenos, que conduce a la supresión prolongada de la secreción hipotalámica de la hormona liberadora de ACTH.
Mecanismos causales
Insuficiencia suprarrenal primaria
En los países desarrollados, el 80–90% de los casos de ISP están ocasionados por la adrenalitis autoinmune, la cual puede ser aislada (40%) o formar parte del síndrome de poliendocrinopatía autoinmune (SPEA) (60%).
La enfermedad de Addison autoinmune se caracteriza por la destrucción de la corteza suprarrenal por mecanismos inmunológicos mediados por células. En el 85% de los pacientes con ISP idiopática se detectan anticuerpos contra el esteroide 21-hnidroxilasa pero esa detección es muy rara en las otras causas.
Por otra parte, en la enfermedad de Addison autoinmune se han identificado otros antígenos, como el esteroide 17α-hidroxilasa y la enzima de escisión de la cadena lateral del colesterol. El mismo hallazgo ocurre en la insuficiencia ovárica primaria.
Las células T y la inmunidad celular también tienen un papel importante en la patogénesis de la enfermedad de Addison autoinmune y la generación de autoanticuerpos puede ser secundaria a la destrucción tisular. Por otra parte, se han identificado varios genes que confieren susceptibilidad a la enfermedad de Addison autoinmune.
La ISP también puede presentarse en el contexto del síndrome de poliendocrinopatía autoinmune (SPEA), cuyos tipos 1 y 2 tienen una base genética. Los anticuerpos contra el interferón-ω y el interferón-α son sensibles y específicos de ese síndrome; el análisis de las mutaciones genéticas confirma el diagnóstico en el 95% de los casos.
El SPEA tipo 2 se caracteriza por la IS autoinmune y la enfermedad tiroidea autoinmune, con o sin diabetes tipo 1; es más prevalente que el tipo 1. Suele asociarse con otras enfermedades autoinmunes, afecta más comúnmente a los hombres y generalmente se presenta en la cuarta década de la vida.
El SPEA tipo 4 es un síndrome raro que se caracteriza por la asociación de la enfermedad de Addison autoinmune con uno o más componentes menores de otras enfermedades autoinmunes (por ej., hipogonadismo, gastritis atrófica, anemia perniciosa, enfermedad celíaca, miastenia grave, vitíligo, alopecia e hipofisitis) pero está excluido el componente mayor del SPEA tipo 1 y 2 (candidiasis crónica, hipoparatiroidismo, enfermedad tiroidea autoinmune y diabetes tipo 1).
Existen otras causas de IS como las infecciosas, la inducción por fármacos, varias mutaciones genéticas entre las que se puede mencionar a la adrenoleucodistrofia, la cual presenta trastornos neurológicos provocados por la desmielinización de la sustancia blanca y las manifestaciones de la ISP, que se presentan en la infancia o la niñez.
Las dos formas principales de adrenoleucodistrofia son la forma cerebral (50% de los casos; se manifiesta en la primera infancia con una rápida progresión) y la adrenomieloneuropatía (35% de los casos, de comienzo en la primera infancia con lenta progresión), en la cual la desmielinización está restringida a la médula espinal y los nervios periféricos. Dado que la manifestación inicial puede ser la IS, hay que sospechar la adrenoleucodistrofia en los varones jóvenes con IS.
Síndrome antifosfolípido: En ocasiones, la ISP se presenta en forma aguda como consecuencia de una hemorragia suprarrenal bilateral en pacientes con síndrome antifosfolípidos. Se caracteriza por trombosis arterial y venosa recurrente, complicaciones gestacionales y autoanticuerpos antifosfolípidos. Puede presentarse aislada o manifestarse en el contexto de trastornos del tejido conectivo o malignos.
Niños: En los niños, la causa más común de ISP es la hiperplasia suprarrenal congénita, un grupo de trastornos recesivos autosómicos ocasionados por la deficiencia de una de las enzimas necesarias para la síntesis del cortisol en la corteza suprarrenal.
La forma más común es la clásica deficiencia de 21-hidroxilasa, la que se caracteriza por la poca síntesis de glucocorticoides y en muchos casos de mineralocorticoides, hiperandrogenismo suprarrenal y alteración del desarrollo y funcionamiento de la médula suprarrenal. Existen otras formas más raras por deficiencia de otras hidroxilasas y enzimas (3β-hidroxiesteroide deshidrogenasa o P450 óxido reductasa).
Insuficiencia suprarrenal central
La IS secundaria es provocada por cualquier proceso que afecte a la hipófisis e interfiera con la secreción de ACTH. La deficiencia de ACTH puede ser aislada o estar asociada a las deficiencias de otras hormonas hipofisarias.
La IS secundaria asilada puede estar causada por un proceso autoimmune, y con frecuencia se asocia a otros trastornos endocrinos autoinmunes (tiroiditis, diabetes tipo 1). La deficiencia de ACTH también puede tener su origen en mutaciones genéticas.
La IS terciaria resulta de procesos que afectan al hipotálamo e interfieren la secreción de la hormona liberadora de ACTH, la vasopresina arginina o ambas. La causa más común de supresión del eje hipotálamo-hipófisis-suprarrenal es la administración prolongada de dosis elevadas de glucocorticoides.
Para lograr la recuperación total de dicho eje, en la mayoría de los casos se requiere la lenta disminución de las dosis de glucocorticoides durante 9–12 meses.
La IS terciaria también ocurre en pacientes que se han recuperado del síndrome de Cushing, ya que las concentraciones persistentemente elevadas de cortisol previas al tratamiento inhiben al eje hipotálamo-hipófisis-suprarrenal, de la misma manera que lo hacen los glucocorticosteroides exógenos.
Por último, los fármacos como la mifepristona, un antagonista del receptor de glucocorticoides, los antipsicóticos y los antidepresivos causan resistencia tisular, debido al deterioro de la señal de transducción de glucocorticoides.
Fisiopatología y presentación clínica
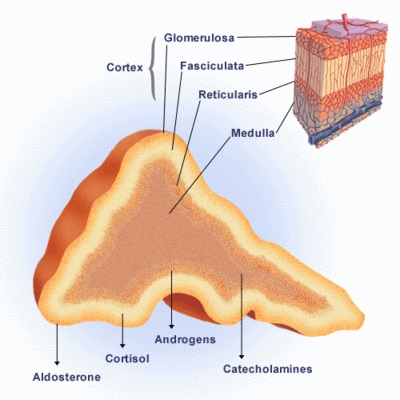 La corteza suprarrenal tiene 3 zonas diferentes, las cuales secretan varias hormonas, bajo el control directo de mecanismos de retroalimentación bien conocidos.
La corteza suprarrenal tiene 3 zonas diferentes, las cuales secretan varias hormonas, bajo el control directo de mecanismos de retroalimentación bien conocidos.
La aldosterona es sintetizada en la zona más externa, la zona glomerular. Su secreción está sobre todo regulada por el sistema renina-angiotensina y las concentraciones de potasio extracelular. Por lo tanto, no está alterada en la IS secundaria o terciaria.
La secreción de cortisol de la zona fasciculada está principalmente regulada por la ACTH, la cual es liberada de la hipófisis anterior en respuesta a la hormona liberadora de ACTH y la vasopresina arginina.
En las personas sanas, la secreción de cortisol es pulsátil y las concentraciones del cortisol circulante fluctúan naturalmente en forma circadiana, con concentraciones más elevadas a la mañana (06:00–08:00 horas) y más bajas alrededor de la medianoche.
Los andrógenos suprarrenales dehidroepiandrosterona y sulfato éster de dehidroepiandorsterona son sintetizados en la zona reticular, más interna. La dehidroepiandrosterona y su sulfato tienen un patrón particular: están relacionados con la edad en el período neonatal las concentraciones son muy elevadas y luego declinan hasta valores muy bajos durante los primeros meses de vida; posteriormente comienzan a aumentar continuamente entre los 6 y los 10 años, lo que se denomina adrenarquia.
Las concentraciones máximas de esas dos hormonas se alcanzan durante la tercera década de la vida; luego declinan sostenidamente desde la quinta década (adrenopausia) hasta que alrededor de los 70 años disminuyen un 10–20% el valor de la concentración máxima.
La disminución del sulfato de dehidroepidandrosterona relacionada con la edad no refleja la pérdida general de la secreción adrenocortical porque las concentraciones de cortisol incluso aumentan ligeramente con la edad.
Manifestaciones clínicas
Las manifestaciones clínicas de la ISP se deben a la deficiencia de todas las hormonas adrenocorticales (aldosterona, cortisol, andrógenos); también pueden observarse signos de otras condiciones autoinmunes concurrentes. La mayoría de los síntomas son inespecíficos y pueden retrasar el diagnóstico y el tratamiento.
En los niños con IS puede haber hipoglucemia y un mal control de la glucosa; en los pacientes con diabetes tipo 1 aparece la necesidad de reducir la dosis total diaria de insulina.
Un signo específico de ISP crónica que no aparece en la ISP aguda es la hiperpigmentación, la cual afecta sobre todo las áreas de piel sujetas a presión (codos, nudillos, pliegues palmares, labios, mucosa oral). Está causada por la estimulación de los receptores cutáneos de la melanocortina-1 debido a las elevadas concentraciones de ACTH circulante.
El período preclínico puede durar muchos años a partir de la detección de los primeros cambios metabólicos, aun en presencia de títulos elevados de autoanticuerpos específicos y concentraciones muy elevadas de ACTH.
En la IS autoinmune, la primera zona afectada por la destrucción inmunitaria suele ser la zona glomerular, quizás debido a que es más delgada que las otras dos zonas, o porque es más vulnerable al ataque autoinmune.
Este cuadro explicaría el primer período de la IS caracterizado por una elevada actividad de la renina plasmática y concentraciones bajas de aldosterona, seguido por una fase de deficiencia progresiva de glucocorticoides, inicialmente con una respuesta inadecuada al estímulo estresante y luego por una fase de insuficiencia manifiesta con concentraciones bajas de cortisol basal.
Las manifestaciones clínicas de la IS secundaria o terciaria resultan solamente de la insuficiencia de glucocorticoides (la secreción de aldosterona y andrógenos suprarrenales está preservada); sin embargo, también puede haber signos de un trastorno primario subyacente.
La hiperpigmentación no está presente porque la secreción de ACTH no está aumentada; secundariamente, puede haber hiponatremia y expansión de volumen debidas al aumento inapropiado de la secreción de vasopresina arginina.
También podría haber síntomas y signos de deficiencia de otras hormonas de la hipófisis anterior. La primera presentación de la IS podría ser una crisis suprarrenal con peligro de muerte. Las manifestaciones clínicas incluyen:
- vómitos
- dolor abdominal
- mialgias
- artralgias
- hipotensión grave
- shock hipovolémico.
La presentación aguda puede ser precipitada por un estrés fisiológico como la cirugía, el trauma o una infección intercurrente.
Diagnóstico de la insuficiencia suprarrenal
Para el diagnóstico de IS existen 3 objetivos principales a confirmar:
- La secreción inapropiadamente baja de cortisol.
- La distinción entre IS primaria o central.
- La identificación de los procesos patológicos subyacentes.
Cualquiera sea la causa, el diagnóstico de IS depende enteramente de demostrar que la secreción de cortisol está inapropiadamente baja. Todas las pruebas de estimulación actuales miden la concentración total de cortisol, la cual está relacionada estrechamente con el cortisol libre biológicamente activo, pero quizás no sea ésta toda la situación.
Las concentraciones elevadas de globulina ligada al cortisol en las pacientes que reciben estrógenos orales durante el embarazo pueden provocar resultados normales falsos. Por el contrario, en los pacientes con cirrosis, las concentraciones de globulina ligada al cortisol son bajas y pueden dar lugar a resultados anormales falsos.
La medición de las concentraciones séricas de cortisol libre puede ofrecer información adicional, aunque en general, los análisis disponibles, como la concentración de cortisol salival, podrían ser una alternativa útil.
En las personas sanas, las concentraciones séricas de cortisol son más elevadas a la mañana temprano (100–200 μg/L).
Una concentración sérica de cortisol baja (<30 μg/L) en una muestra de sangre a la mañana temprano es un fuerte indicador d IS.
Por el contrario, en la mayoría de los pacientes, una concentración sérica de cortisol matinal >150 μg/L supone una respuesta normal del cortisol sérico a la hipoglucemia inducida por la insulina, o a una prueba de ACTH corta. La medición simultánea de las concentraciones de cortisol y ACTH identifica a la mayoría de los casos de ISP.
Del mismo modo, la concentración de cortisol salival >5,8 μg/L a las 08:00 horas excluye la IS, mientras que un valor <1,8 μg/L indica una alta probabilidad de IS. Esta prueba se ha utilizado como prueba de detección para la IS pero no ha sido completamente validada como prueba diagnóstica.
La medición de las concentraciones de cortisol libre en la orina no es útil para el diagnóstico de IS, principalmente porque los niveles más bajos del rango no son contributivos.
En general, la medición de la concentración de ACTH plasmática basal distingue la ISP de la IS central. La medición simultánea de la concentración de cortisol sérico basal y de la ACTH plasmática puede confirmar el diagnóstico de IS y establecer su causa.
En las personas sanas, las concentraciones de ACTH a las 08:00 horas son de 20–52 ng/L.
En la ISP, la concentración de ACTH plasmática a las 08:00 horas es elevada y se asocia con concentraciones o elevadas o mayor actividad de la renina plasmática, concentraciones bajas de aldosterona, hiponatremia e hiperpotasemia.
Por el contrario, en la IS secundaria o terciaria, la concentración de ACTH plasmática es baja o normal baja y las concentraciones plasmáticas de renina y aldosterona suelen estar inalteradas, pero luego de una duración muy prolongada de deficiencia de ACTH aparece una deficiencia de mineralocorticoides.
Habitualmente, las primeras concentraciones que aumentan en la IS autoinmune son las de la renina plasmática seguidas por el aumento de la concentración de ACTH y el descenso de la concentración de aldosterona.
La IS se caracteriza por la deficiencia de andrógenos suprarrenales. En los pacientes con ISP o central, las concentraciones séricas de dehidroepiandrosterona y su sulfato son bajas, pero estas características facilitan el diagnóstico solamente en los pacientes <40 años, debido a la fisiología y a la declinación de los andrógenos suprarrenales relacionadas con la edad.
En general, la IS se diagnostica mediante la prueba de ACTH con dosis estándar, la cual es segura, confiable y precisa.
Por medio de esta prueba se estimulan las glándulas suprarrenales con dosis farmacológicas (250 μg) de ACTH 1-24 exógena, la cual tiene toda la potencia biológica del aminoácido-39 de la ACTH. Se administra por vía intravenosa o intramuscular y se miden las concentraciones de cortisol sérico al comienzo y a los 30 y 60 minutos después de la administración.
La concentración pico normal de cortisol es >180 μg/L. La prueba de ACTH con dosis estándar no debe ser usada durante las primeras 4–6 semanas después de un insulto hipotalámico o hipofisario porque la corteza suprarrenal podría todavía estar respondiendo adecuadamente a la administración de la ACTH exógena y dar un resultado normal falso.
Debido a que algunos pacientes con IS pueden tener una respuesta de cortisol normal a la prueba con dosis estándar de estimulación, dado que la dosis farmacológica de la hormona es suficiente para suscitar una respuesta, se han investigado pruebas con dosis más bajas (1 µg/L o 500 mg/1,73 m2) para aumentar la sensibilidad.
Esta prueba puede ser más sensible y específica que la prueba estándar. Pero hay detalles técnicos que pueden influir en su precisión. Por lo tanto, la prueba se reservará para los pacientes con ISP o IS central de reciente comienzo (4-6 semanas de duración).
La estimulación prolongada con ACTH exógena se usa para diferenciar la IS primaria, secundaria y terciaria. En la ISP, las glándulas suprarrenales no responden a la ACTH, mientras que en las otras dos, la respuesta aparece luego de períodos más prolongados de estimulación con ACTH.
La prueba implica la administración de 250 μg de ACTH por infusión intravenosa durante 24 horas, 2 o 3 días consecutivos, y se mide la concentración de cortisol sérico y de cortisol en la orina de 24 horas, como así la concentración de 17-hidroxicorticoide, antes y después de la infusión.
Ante la sospecha de IS secundaria, otra opción para confirmar el diagnóstico es hacer la prueba de tolerancia a la insulina, particularmente en los pacientes con sospecha de deficiencia de ACTH de reciente comienzo. Esta prueba investiga la integridad del eje hipotálamo-hipófisis-suprarrenal y es ampliamente aceptada como el estándar de oro.
Por otra parte, tiene la ventaja de evaluar la reserva de hormona de crecimiento. No obstante, no se debe hacer en los pacientes con enfermedad cardiovascular o antecedente de convulsiones y requiere una supervisión muy estrecha. Se administran 0,10–0,15 U/kg de insulina para inducir hipoglucemia y luego medir las concentraciones de cortisol cada 30 minutos, durante al menos 120 minutos.
La prueba de la hormona liberadora de ACTH evalúa la reserva de ACTH hipofisaria. Esta prueba puede ser útil para distinguir la IS secundaria de la terciaria, aunque esta diferenciación raramente es importante para el tratamiento y requiere la administración intravenosa de la hormona liberadora de ACTH a una dosis de 1 μg/kg (hasta un máximo de 100 μg) y la medición de las concentraciones de cortisol sérico y de ACTH plasmática basal, cada 15 minutos hasta 1 hora después de la estimulación y luego, cada 30 minutos hasta las 2 horas.
En los pacientes con IS secundaria no hay respuesta a la ACTH o es muy escasa, mientras que en los pacientes con IS terciaria hay una respuesta exagerada y prolongada a esa hormona.
Los autores llaman la atención de que ninguna de esas pruebas dinámicas, incluyendo el test de tolerancia a la insulina, clasifica correctamente a los pacientes con IS.
La IS secundaria leve puede pasar desapercibida y los individuos sanos pueden mostrar respuestas ligeramente anormales. Por lo tanto, es muy importante el criterio clínico, y los pacientes con síntomas persistentes sugestivos de IS deben volver a evaluarse.
El diagnóstico de IS autoinmune se basa en el hallazgo de autoanticuerpos contra la corteza suprarrenal, sin agrandamiento de las suprarrenales en la radiografía (tamaño normal o pequeño) y la presencia de otras enfermedades autoinmunes, con la exclusión de las otras causas conocidas de IS.
En más del 90% de los pacientes con adrenalitis autoinmunes de reciente comienzo se hallan autoanticuerpos anti corteza suprarrenal, o anti 21-hidroxilasa. Por otra parte, en algunos pacientes hay autoanticuerpos contra otras enzimas esteroideogénicas (P450scc, P450c17) y contra las células productoras de esteroides, los que pueden ser marcadores predictivos de insuficiencia ovárica primaria.
En los pacientes varones con enfermedad de Addison aislada y sin autoanticuerpos, se deben medir las concentraciones plasmáticas de los ácidos grasos de cadena muy larga, para excluir la adrenoleucodistrofia ligada a X.
Los pacientes sin una enfermedad autoinmune ni autoanticuerpos deben ser estudiados mediante una tomografía computarizada (TC) de las glándulas suprarrenales. En los países en desarrollo y poblaciones de inmigrantes, un diagnóstico diferencial es la adrenalitis tuberculosa.
En las primeras etapas, la TC muestra la hiperplasia de las glándulas suprarrenales, y en los estadios posteriores de la enfermedad calcificaciones irregulares. Otras causas más raras de IS que pueden ser detectadas mediante la TC suprarrenal son el linfoma suprarrenal bilateral, las metástasis suprarrenales o la infiltración suprarrenal ((sarcoidosis, amiloidosis, hemocromatosis).
Ante la sospecha de IS central, están indicadas las imágenes por Resonancia Magnética de las regiones hipotalámica e hipofisaria, ya que pueden revelar adenomas hipofisarios, craneofaringiomas, meningiomas, metástasis e infiltración sarcoidótica, histiocitosis de células de Langerhans u otras enfermedades granulomatosas.
Las imágenes no son necesarias en presencia de anticuerpos contra la corteza suprarrenal.
Tratamiento
La IS puede poner en peligro la vida. El tratamiento debe iniciarse tan pronto como se confirma el diagnóstico o más pronto aún si el paciente presenta en crisis suprarrenal.
|
Prevención y manejo de la insuficiencia suprarenal Insuficiencia suprarrenal aguda |
Una parte muy importante del manejo de la IS crónica es la educación del paciente y su familia, quienes necesitan conocer la importancia de la terapia de reemplazo para toda la vida, la necesidad de aumentar la dosis usual de corticosteroides durante el estrés y de notificar al equipo médico si el paciente está por ser sometido a un procedimiento quirúrgico.
Por otra parte, ellos siempre deben tener a su alcance inyecciones de hidrocortisona y saber cómo y cuándo administrarla.
Los pacientes con IS deben ser tratados con hidrocortisona (o cortisona cuando no hay disponibilidad de acetato de hidrocortisona), que es la opción más fisiológica para el reemplazo glucocorticoide. La dosis diaria recomendada de hidrocortisona es 10–12 mg/m²; puede darse en 2-3 dosis administrando la mitad o los dos tercios de la dosis total diaria a la mañana.
En los pacientes con tratamiento prolongado con hidrocortisona se pueden observar descensos leves de la densidad mineral ósea. Hay que evitar los glucocorticoides sintéticos de acción prolongada como la prednisona y la dexametasona porque la larga duración de su acción puede provocar signos de exceso crónico de glucocorticoides.
Últimamente se han desarrollado preparaciones de hidrocortisona de liberación retardada y sostenida que están bajo investigación clínica. Estas formulaciones brindan concentraciones de cortisol más estables a lo largo de los días y reproducen la elevación fisiológica del cortisol en las primeras horas de la mañana, después de la ingesta oral de la preparación al acostarse. También se ha desarrollado un comprimido de hidrocortisona de liberación dual de una sola toma diaria para obtener un perfil de exposición al cortisol sérico con un ritmo circadiano más fisiológico.
Comparado con el enfoque tradicional, el tratamiento con esta preparación mejoró los factores de riesgo cardiovasculares, el metabolismo de la glucosa y la calidad de vida, todo lo cual puede ayudar a mejorar el resultado en los pacientes con IS.
En ausencia de variables objetivas para evaluar si la terapia de reemplazo es adecuada, el médico tiene que confiar principalmente en los síntomas y signos que indican que el reemplazo de glucocorticoides es bajo o excesivo, para optar por titular apropiadamente la dosis y prevenir una morbilidad significativa.
Durante enfermedades o procedimientos quirúrgicos menores, la dosis de glucocorticoide puede aumentarse hasta triplicar la dosis usual de mantenimiento. Durante la enfermedad o cirugía mayor, dicho aumento podría llegar a 10 veces, para evitar la crisis suprarrenal.
En la ISP es necesaria la terapia de reemplazo mineralocorticoide, para prevenir la pérdida de sodio, la depleción de volumen intravascular y la hiperpotasemia. Se administra en forma de glucocortisona (9-α-?uorohidrocortisona), en una dosis de 0,05–0,20 mg/día, a la mañana. La dosis de fluorocortisona es monitoreada individualmente mediante el control de la presión arterial, la natremia y la potasemia, como así de las concentraciones de la actividad de la renina plasmática.
La dosis de mineralocorticoide tendría que ser aumentada en el verano, especialmente si los pacientes están expuestos a temperaturas >29°C. En la IS secundaria o terciaria no es necesario el reemplazo de mineralocorticoides, pero sí podrían serlo en las deficiencias de la hipófisis anterior.
En las mujeres, la corteza suprarrenal es la fuente principal de producción de andrógenos, como hidroepiandrosterona y su sulfato. En los pacientes adultos, niños y adolescentes con IS, el tratamiento con dehidroepiandrosterona mejora el humor y la sensación de bienestar general.
El reemplazo de la dehidroepiandrosterona está indicado en los pacientes cuyo bienestar está muy alterado a pesar de recibir las dosis óptimas de reemplazo de glucocorticoides y mineralocorticoides. Una sola dosis oral de 25-50 mg a la mañana es suficiente para mantener las concentraciones séricas dentro de los límites normales.
La vigilancia terapéutica incluye la medición de las concentraciones séricas del sulfato de dehidroepiandrosterona (valor objetivo: la media del rango normal para las personas jóvenes sanas) y las mujeres deben saber que deben informar cualquier efecto androgénico.
El objetivo terapéutico de la hiperplasia suprarrenal congénita clásica no solo es proveer un reemplazo adecuado de glucocorticoides y mineralocorticoides para evitar la crisis suprarrenal sino también suprimir la secreción excesiva de ACTH de la hipófisis anterior, con el fin de disminuir el exceso de producción de andrógenos suprarrenales.
En la niñez, para el control satisfactorio de la secreción de andrógenos suprarrenales se administran 10–15 mg/m² de hidrocortisona diaria en 3 dosis pero en el período neonatal podrían requerirse dosis más elevadas. Dado que estas dosis superan los niveles de la secreción fisiológica de cortisol, los pacientes deben ser monitoreados cuidadosamente para detectar signos del síndrome de Cushing.
La eficacia terapéutica se evalúa mediante la velocidad del crecimiento, la maduración esquelética, la ganancia de peso y la concentración sérica de 17-hidroxiprogesterona y androstendiona a las 08:00 horas. En los pacientes con hiperplasia suprarrenal clásica con alteración de la secreción de aldosterona está indicado el reemplazo mineralocorticoide.
La crisis suprarrenal es una emergencia que puede provocar la muerte y ocurre frecuentemente en los pacientes con IS que reciben terapia de reemplazo estándar, y requiere el tratamiento inmediato. El manejo inicial de la crisis suprarrenal es tratar la hipotensión y revertir las anormalidades electrolíticas y la deficiencia de cortisol.
El tratamiento consiste en la administración inmediata de 100 mg de hidrocortisona y la rehidratación rápida con solución fisiológica normal con monitoreo cardíaco continuo, seguida de 100–200 mg de hidrocortisona en glucosa al 5% durante 24 horas, por infusión intravenosa continua; como alternativa, la hidrocortisona puede darse por inyección intravenosa o intramuscular, cada 6 horas, en dosis de 40-100 mg, dependiendo de la edad y el área de la superficie corporal.
En la ISP, si el paciente recibe ≥50 mg diarios de hidrocortisona, se puede suspender o reducir el reemplazo de mineralocorticoide, porque esa dosis es equivalente a 0,1 mg de ?udrocortisona. Una vez que el paciente se ha estabilizado, el tratamiento con glucocorticoides intravenosos se puede ir disminuyendo durante los días siguientes y ser sustituido por una dosis de mantenimiento oral.
Diagnóstico especial y condiciones terapéuticas
Insuficiencia suprarrenal en pacientes críticos
La IS es común en los pacientes críticamente enfermos y se informa cada vez más en la sepsis, la neumonía grave, el síndrome de distrés respiratorio del adulto, el trauma, la infección por el VIH o después del tratamiento con etomidato.
También puede asociarse con el daño estructural de la glándula suprarrenal, la hipófisis o el hipotálamo. Sin embargo, muchos pacientes críticamente enfermos desarrollan una insuficiencia del eje hipotálamo-hipófisis-suprarrenal reversible.
El mecanismo fisiopatológico que lleva a la IS en el curso de una enfermedad crítica no está bien establecido. No obstante, se mencionan la disminución de la secreción de cortisol y la alteración de la señal de transducción de los glucocorticoides. Se ha postulado que las citocinas proinflamatorias podrían competir con la ACTH en sus receptores o inducirían la resistencia tisular a los glucocorticoides.
En ausencia de glucocorticoides, sus receptores están principalmente en el citoplasma integrando un complejo con las proteínas del shock térmico y las inmunofilinas. Una vez producida la unión de los ligandos, los receptores son sometidos a cambios en su conformación y se separan de las proteínas chaperonas para moverse dentro de los núcleos donde se unen a elementos de la respuesta glucocorticoide negativa o positiva, en las regiones promotoras de genes antiinflamatorios o proinflamatorios.
Por lo tanto, el receptor de glucocorticoides puede inhibir la expresión de genes proinflamatorios, independientemente de la unión del ADN, por la interacción física con el factor de transcripción p65, una subunidad del factor nuclear ?B. Los glucocorticoides pueden inducir efectos antinflamatorios a través de vías no genómoicas. Los receptores de glucocorticoides unidos a la membrana pueden activar las vías de las cinasas en cuestión de minutos.
La activación de la vía MAPK provoca la inhibición de la fosfolipasa citosólica A2α, mientras que la fosfatidilinositol 3-cinasa activada induce la óxido nítrico sintetasa endotelial y la producción subsiguiente de óxido nítrico.
Por otra parte, los glucocorticoides pueden perjudicar la señalización de los receptores de las células T mediante la inhibición no genómica de la cinasa relacionada con el concogen FYN-y, la proteína tirosinacinasa específica de los linfocitos en el receptor de glucocorticoides.
Además de la sepsis por sí misma, los medicamentos usados durante su tratamiento pueden interferir en la síntesis de glucocorticoides y la señal de transducción.
Por otra parte, la alteración del flujo sanguíneo de la hipófisis distal pude provocar isquemia o necrosis hipofisaria, mientras que la creciente acumulación de óxido nítrico, superóxido o, neuropéptidos o prostaglandinas centrales puede contribuir al descenso de la secreción de hormonas hipotálamo-hipofisarias en los pacientes con sepsis.
Un estudio multidisciplinario de pacientes críticos concluyó que en este contexto, el diagnóstico se confirma cuan el cortisol sérico total es <90 μg/L luego de la administración de 250 μg de ACTH, o por una medición al azar de cortisol total <100 μg/L.
Para el shock séptico se recomienda la administración de 200 mg de hidrocortisona diarios, divididos en 4 dosis o en infusión continua de of 240 mg/día (10 mg/h) durante al menos 7 días la. Para los pacientes con el síndrome de disstrés respiratorio del adulto precoz grave se recomienda la metilprednisolona, en dosis de 1 mg/kg/día durante al menos 14 días. El papel del tratamiento glucocorticoide en otros pacientes críticamente enfermos no ha sido del todo estudiado.
Disfunción tiroidea
En los pacientes con IS e hipertiroidismo no tratado, la dosis de glucocorticoides de reemplazo debe ser 2-3 veces superior a la usual, con el fin de compensar la mayor depuración de cortisol que ocurre en el hipertiroidismo. Por otra parte, para prevenir la crisis suprarrenal, la terapia de reemplazo con tiroxina en el hipotiroidismo debe comenzar luego de haber excluido o tratado la IS.
Embarazo
La IS en el embarazo es bastante rara pero si no se diagnostica y trata oportunamente puede provocar una gran morbilidad y también la muerte, tanto de la madre como del feto. El embarazo es un estado fisiológico de exceso de glucocorticoides y se asocia con concentraciones elevadas de globulina ligada al cortisol, al cortisol libre y a la progesterona, especialmente en las últimas semanas de gestación.
Las pacientes con IS en el embarazo deben ser tratadas con 12–15 mg/m² de hidrocortisona diarios, divididos en 3 dosis desiguales, la más concentrada a la mañana. Al comienzo del trabajo de parto, la dosis diaria de hidrocortisona se aumenta al doble o al triple o se administra una dosis parenteral de 50–100 mg, la cual puede ser administrada durante la segunda etapa del trabajo de parto. Si el parto es por cesárea, la hidrocortisona se debe administrar por vía intravenosa en dosis de 100 mg/6 horas, para luego ir disminuyendo en las 48 horas siguientes.
Interacciones medicamentosas
Los anticonvulsivos como la fenitoína, el fenobarbital y la carbamazepina estimulan el citocromo P450 3A4 y de esta manera estimulan las enzimas hepáticas y provocan un metabolismo acelerado de los glucocorticoides, con reducción de su efecto. Por el contrario, los fármacos antirretrovirales, como el ritonavir, inhiben la actividad del citocromo P3A y provocan un retardo en el metabolismo de los glucocorticoides y el consiguiente aumento de su concentración.
♦ Traducción y resumen objetivo: Dra. Marta Papponetti