Los síntomas leves de sangrado son bastante comunes en los niños y no siempre indican un trastorno hemorrágico subyacente. La epistaxis y la fácil aparición de hematomas se han reportado en el 39% y el 24% de los niños, respectivamente. La fácil aparición de hematomas puede estar relacionada con la actividad "normal" de la niñez y su prevalencia varía según la etapa del desarrollo motor, con reporte de hematomas en el 61% al 90% de los niños de 2 años a 10 años de edad. Otros factores que causan sangrado en los niños incluyen anormalidades anatómicas que contribuyen a eventos de sangrado grave, tales como hemorragia intracraneal, gastrointestinal, o genitourinaria, y sangrado menstrual abundante en adolescentes resultante de la inmadurez del eje hipotálamo-hipofisario-ovárico y anovulación. La tasa combinada de sangrado después de una amigdalectomía, un procedimiento pediátrico común, ha sido reportada como del 3,3% en los pacientes sin trastornos de sangrado.
Por lo tanto, la identificación de un niño con sangrado patológico puede resultar difícil, dada la frecuencia relativa de sangrado no patológico y las similitudes entre el sangrado patológico y no patológico. Las áreas comunes de sangrado (por ejemplo, la piel y las membranas mucosas) son similares, y considerando que el sangrado ocurre a menudo espontáneamente en los trastornos hemorrágicos, los individuos afectados pueden inicialmente presentar sangrado sólo después de un trauma o una cirugía, o, en las niñas, con la menarca. A la inversa, los niños con trastornos hemorrágicos pueden tolerar ciertos desafíos hemostáticos sin aparente sangrado excesivo, sólo para presentarse con sangrado patológico más tarde en la vida. Los factores que pueden ayudar a diferenciar el sangrado patológico del no patológico incluyen la edad al primer episodio, la frecuencia, extensión y duración del sangrado, la historia personal de sangrado espontáneo recurrente o relacionado con procedimientos o sangrado menstrual abundante en las niñas, especialmente si se acompaña de otro sangrados, y los antecedentes familiares de sangrado o de diátesis hemorrágica conocida (Tabla 1). El trauma no accidental también debe ser considerado en niños con síntomas de sangrado inexplicables o excesivos. Específicamente, cualquier contusión en un niño sin movilización debe ser vista como patológica y puede indicar ya sea un trastorno hemorrágico o un trauma no accidental. En los niños que deambulan, los hematomas fuera de las prominencias óseas, particularmente en la cara, cabeza o cuello, son especialmente sugerentes de trauma no accidental.
El sangrado patológico se produce en el entorno de anormalidades congénitas o adquiridas en la hemostasia normal, que consiste en 3 procesos básicos: la hemostasia primaria, que culmina con la formación de un tapón plaquetario; la hemostasia secundaria, que comprende los procesos de coagulación (es decir, la formación de fibrina) y la estabilización del coágulo; y la fibrinólisis. Los trastornos hemostáticos primarios que afectan a los niños, a saber, las anormalidades cualitativas y cuantitativas de las plaquetas y del factor de von Willebrand, se revisan en otro lugar, como los trastornos de fibrinólisis excesiva.
Los trastornos hemostáticos secundarios abarcan las deficiencias congénitas o adquiridas de los factores de la coagulación. Entre los trastornos congénitos, la hemofilia A (HA) y la hemofilia B (HB) son los más comunes, que resultan de las deficiencias del factor VIII (FVIII) y el factor IX (FIX), respectivamente. La HA y la HB se heredan principalmente como trastornos recesivos ligados al cromosoma X y juntos afectan ~ 1 cada 5000 varones nacidos vivos por año en los Estados Unidos, correspondiendo a HA ~ el 80% de los casos. Las características clínicas de la HA y la HB se basan en el sangrado musculo-esquelético y de tejidos blandos, el primero de los cuales puede conducir a una artropatía debilitante. En la HA y la HB, el riesgo de sangrado y el fenotipo se correlacionan con los niveles plasmáticos del factor deficiente en la mayoría de los casos.
Las deficiencias heredadas de otros factores de la coagulación que pueden manifestarse con hemorragias ocurren con mucha menor frecuencia y por lo tanto se hace referencia a las mismas colectivamente como trastornos de la coagulación "raros" (TCRs). Los TCRs consisten en deficiencias heredadas cuantitativas o funcionales de los factores I (es decir, fibrinógeno [FI]), II (FII), V (FV), VII (FVII), X (FX), XI (FXI) y XIII (FXIII) y deficiencias combinadas de factores, más notablemente de FV y FVIII y de factores vitamina K dependientes. Dada la naturaleza congénita de estas condiciones, se pueden presentar con sangrado en la lactancia o la infancia. Aunque los TCRs graves pueden presentarse inicialmente con complicaciones por sangrado severo como hemorragia intracraneal (HIC), tales eventos pueden ser ocasionalmente anunciados por síntomas menos graves. El reconocimiento de las características históricas y de laboratorio que sugieren la presencia de un TCR es de suma importancia para optimizar la rapidez en la consulta con un hematólogo para diagnóstico definitivo, tratamiento, y seguimiento a largo plazo de los lactantes y niños afectados. Sin embargo, la identificación de estos trastornos es desafiada por su rareza, sus variables y a menudo indistinguibles presentaciones clínicas, y la frecuente falta de antecedentes familiares para sugerir un trastorno de la coagulación hereditario. Mientras que se sabe mucho (y se ha escrito) sobre la presentación clínica y el manejo de la HA y HB en niños, la información similar relativa a los TCRs es relativamente limitada en general, así como en la literatura pediátrica, en gran parte debido al pequeño número de niños identificados con estos trastornos. Esta revisión resume la presentación clínica, diagnóstico y manejo básico de los TCRs en niños para pediatras no hematólogos, con énfasis en un enfoque de atención primaria.
Tabla 1. Características iniciales sugestivas de sangrado patológico en niños
- Hematomas múltiples y palpables en lactantes y niños mayores sin movilización independiente - Hematomas palpables persistentes en un niño mayor deambulador - Sangrado espontáneo en ausencia de causas anatómicas
- Sangrados atípicos (ej. hemartrosis, sangrado retroperitoneal) espontáneos o provocados - Sangrado excesivo o prolongado después de desafíos hemostáticos (ej. trauma, procedimientos dentales, cirugía) - Menorragia en mujeres adolescentes: sangrado menstrual por > 7 días o pérdida de sangre > 80 ml por ciclo menstrual (demostrado al empapar la toalla higiénica o el tampón dentro de la hora o por el cambio de toallas higiénicas o tampones a cada hora, o bien por la eliminación de grandes coágulos [> 1.1 pulgadas de diámetro])
- Sangrado excesivo o prolongado después de un traumatismo o de procedimientos invasivos - Diátesis hemorrágica conocida o presunta
- Hallazgos físicos sugestivos de causas específicas subyacentes (ej. Petequias en trastornos plaquetarios, ictericia en enfermedad hepática, hipermovilidad, malformaciones vasculares, anormalidades músculo-esqueléticas) - Palidez/anemia |
Epidemiología y presentación clínica
Las características que distinguen a los TCRs de los trastornos de la coagulación hereditarios más comunes, la HA y HB, se resumen en la Tabla 2. Los TCRs abarcan un 3% a 5% de todos los trastornos de la coagulación hereditarios en general, y, específicamente, en niños.
Con una prevalencia de 1 persona sintomática por 500.000 habitantes, la deficiencia del FVII es el TCR más común, constituyendo el 30% a 50% de los diversos TCRs incluidos en las series publicadas, seguido por la deficiencia del FXI, que representa el 23% al 39% de los TCRs en diversos registros multinacionales. Los TCRs menos frecuentes son las deficiencias de FII y FXIII, con una prevalencia de aproximadamente 1 por cada 2 millones cada uno, y la deficiencia de factores de la coagulación vitamina K dependientes (DFCKD), reportándose < 30 casos a partir del 2008.
Tabla 2. Características epidemiológicas, genéticas, clínicas y de laboratorio distintivas de las Hemofilias y los TCRs
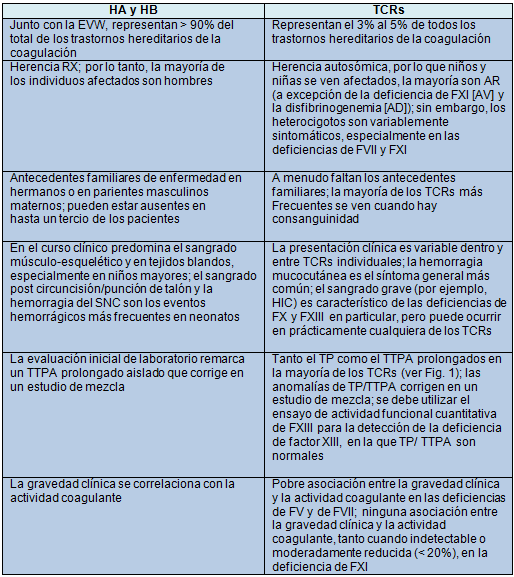
AD, autosómica dominante; AV, autosómica con penetrancia variable; SNC, sistema nervioso central; EVW, enfermedad de von Willebrand; RX, recesiva ligada al X.
Herencia
La mayoría de los TCRs son considerados autosómicos recesivos (AR); sin embargo, en todos menos en la deficiencia combinada de FV/FVIII y en la DFCKD, los heterocigotos (que suman 1 por cada 350 a 700 individuos), pueden tener diversos grados de deficiencia del factor correspondiente que llevan a una impredecible propensión a la hemorragia, especialmente en las deficiencias de FVII y FXI. Mientras que la ocurrencia de heterocigotos sintomáticos contradice el modelo de herencia AR, la prevalencia de la mayoría de estos TCRs se incrementa cuando la consanguinidad es un factor común, similar a otras condiciones AR. Además, a menudo se carece de una historia familiar de sangrado. La deficiencia combinada de FV/FVIII y la DFCKD, que son causadas por mutaciones en los genes fuera de los que codifican los factores de coagulación en sí mismos, son trastornos AR verdaderos en los que los heterocigotos tienen niveles normales de factores y por lo tanto son asintomáticos. Los TCRs estrictamente no-AR incluyen la deficiencia de FXI (autosómica con penetrancia variable) y la disfibrinogenemia (autosómica dominante).
No hay propensiones inherentes basadas en la raza o etnia para los TCRs específicos, con la excepción de la deficiencia de FXI, que es especialmente prevalente entre los Judíos Ashkenazi; 1 de cada 450 se ve afectado y el 8 % son heterocigotos, representando la mayor incidencia de enfermedad grave, en particular, en este grupo. Además, se halló que la deficiencia de FII afecta de manera desproporcionada a los latinos en una encuesta realizada en 58 centros de tratamiento de hemofilia en América del Norte.
Manifestaciones clínicas de los TCRs
Síntomas comunes de sangrado
La propensión al sangrado varía ampliamente entre las personas afectadas por TCRs, desde la ausencia de síntomas (45,8% de 489 personas con TCRs en una serie Europea) a un sangrado potencialmente mortal. Sin embargo, cuando los pacientes son considerados a través de todas las edades y genotipos, la manifestación de sangrado más común de los TCRs en general es el sangrado mucocutáneo. Esta característica puede hacer que sea difícil distinguir clínicamente los TCRs de los trastornos plaquetarios o de la enfermedad de von Willebrand o de una hemorragia no patológica. En contraste con el sangrado no patológico, la hemorragia relacionada con los TCRs es a menudo espontánea. Alternativamente, los TCRs pueden presentarse con sangrado excesivo después de un desafío hemostático. Por ejemplo, el sangrado después de procedimientos como la circuncisión o la punción en el talón en el período neonatal y de extracciones dentales más tarde en la infancia es un síntoma común de todos los TCRs, aunque la ausencia histórica de sangrado después de estos desafíos no excluye necesariamente un TCR.
Síntomas de sangrado específicos por sitio
La presentación de los TCRs individuales puede ser diversa. En general, el sangrado grave (por ejemplo, HIC o sangrado musculo-esquelético o de cordón umbilical) es raro en las deficiencias de FV y FXI, como es el sangrado espontáneo en la deficiencia de FXI. El fenotipo del sangrado es ampliamente heterogéneo en las deficiencias de FV, FVII, y FXI y, en contraste con la HA y HB, se correlaciona pobremente con el nivel de actividad del factor. Específicamente, una reciente revisión documentó una pobre correlación entre la gravedad clínica y la actividad coagulante en las deficiencias de FV y de FVII, y una falta de correlación entre la gravedad clínica y la actividad coagulante en la deficiencia de FXI, incluso a niveles indetectables o moderadamente reducidos (20%). En particular, la deficiencia severa de FVII puede ser asintomática o, por el contrario, puede presentarse con eventos de sangrado graves (por ejemplo, HIC) en la infancia.
En las deficiencias de FI, FII, FX, y FXIII, la gravedad clínica se correlaciona fuertemente con los niveles del factor. El fenotipo de sangrado en la deficiencia de FI (afibrinogenemia) puede ser similar al de la HA o HB moderada a severa; sin embargo, el sangrado del cordón umbilical y el sangrado de la mucosa están entre los síntomas más comunes. La deficiencia severa del FII se presenta a temprana edad con sangrado mucoso y musculo-esquelético y HIC. Los episodios hemorrágicos graves como el sangrado del cordón umbilical, el hemoperitoneo ovulatorio, y la hemartrosis son relativamente comunes en la deficiencia de FII en relación con otros TCRs. Del mismo modo, las deficiencias de FX y de FXIII se caracterizan generalmente por el inicio temprano de episodios de sangrado graves o potencialmente mortales, tales como HIC o sangrado umbilical. Este último es prácticamente patognomónico de la deficiencia de factor XIII y a menudo se retrasa, dado el papel del FXIII en la estabilización de los coágulos.
Aunque más comunes en ciertos TCRs, los episodios hemorrágicos graves pueden ocurrir en prácticamente cualquier TCR y por lo tanto no siempre se puede distinguir un TCR de otro o de otros trastornos de la coagulación. Por ejemplo, el sangrado del cordón umbilical puede ocurrir en otras condiciones distintas a las deficiencias de FX y FXIII, incluyendo HA, HB, y otros TCRs.
Del mismo modo, la HIC se ha descripto en el periodo neonatal en severas deficiencias del FII, FV y FVII y en DFCKD, aunque la HIC es especialmente común en las deficiencias de FX y FXIII; la HIC se ha reportado en hasta el 20% de los individuos con deficiencia del FX y como en hasta un 30% de aquellos con deficiencia de factor XIII, en la que la HIC es la principal causa de muerte y discapacidad. Otros eventos hemorrágicos que amenazan la vida o un miembro, como el hemoperitoneo ovulatorio y la hemartrosis, han sido informados con mayor frecuencia en las deficiencias de FII, FX, y FXIII. La deficiencia de FX se ha asociado con una variedad de anomalías no relacionadas con la coagulación, incluyendo síndrome de ausencia de radio-trombocitopenia, prolapso de la válvula mitral, miocardiopatía hipertrófica e hipercolesterolemia. La deficiencia de FV es a veces confundida con el FV Leiden. La deficiencia de FV es un trastorno hereditario que se caracteriza por bajos niveles de FV debido a mutaciones en el gen FV, que impide la síntesis de esta proteína procoagulante. Por lo tanto, los individuos con deficiencia de FV tienen un mayor riesgo de sangrado excesivo. El FV Leiden, por otro lado, es una trombofilia genética en la que un defecto en el gen FV hace un FV resistente a la inactivación por la proteína C anticoagulante, aumentando así el riesgo de eventos coagulantes como trombosis venosa profunda, embolia pulmonar, y abortos involuntarios.
Sangrado en mujeres con TCRs
La menorragia es el síntoma de sangrado más común en mujeres con trastornos de la coagulación, incluyendo los TCRs. Aproximadamente el 50% de las mujeres afectadas por TCRs reportan menorragia.
La menarca puede ser el primer desafío hemostático significativo en las niñas con TCRs, pero los datos relativos a la prevalencia de TCRs entre adolescentes con menorragia son limitados. La menorragia es probablemente subregistrada en las adolescentes en general y constituye una fuente "oculta" de hemorragia, por lo que es difícil de identificar sin la revelación por parte del paciente o sin el interrogatorio explícito de los clínicos. Factores sugestivos de menorragia se presentan en la Tabla 1. La presencia de lesiones estructurales (por ejemplo, leiomiomas) pueden agravar la menorragia en mujeres con TCRs e incluso pueden llevar a sangrados potencialmente mortales. Las mujeres con TCRs también están en mayor riesgo de desarrollar quistes ováricos hemorrágicos, que son una manifestación menos común pero tal vez más específica de un trastorno hemorrágico subyacente más que de menorragia. Una ruptura de un folículo puede conducir a hemoperitoneo en mujeres con TCRs, como se describió anteriormente. Las mujeres con TCRs pueden experimentar también complicaciones obstétricas, incluyendo hemorragia post-parto y abortos involuntarios recurrentes debido a los roles que juegan los factores deficientes (es decir, FXIII y FI) en la implantación de la placenta y el mantenimiento del embarazo.
Deficiencias combinadas de factores
Aún más raras que las deficiencias aisladas son las deficiencias combinadas de factores. La deficiencia combinada de FV/FVIII se produce debido a un defecto molecular común que afecta el procesamiento intracelular de ambas proteínas, más que a defectos coincidentes en los genes de ambos factores. Esta condición se asocia con un fenotipo de hemorragia leve, sin ningún efecto acumulativo aparente de las deficiencias combinadas.
En contraste, la DFCKD se asocia a menudo con sangrado significativo. La DFCKD resulta de defectos hereditarios en la activación (ɣ-carboxilación) de los factores vitamina K-dependientes (FII, FVII, FIX, y). Los niños afectados suelen presentar en la infancia o en la juventud temprana eventos graves de sangrado, incluyendo HIC. También se han reportado anomalías esqueléticas, posiblemente debido a una ɣ-carboxilación defectuosa de las proteínas de la matriz ósea.
Evaluación diagnóstica y consideraciones
Después de obtener los antecedentes personales y familiares, la evaluación de laboratorio inicial de un niño con sospecha de hemorragia patológica debe consistir en un recuento sanguíneo completo y en estudios de coagulación de rutina (tiempo de protrombina [TP], tiempo de tromboplastina parcial activada [TTPA], FI, y tiempo de trombina). Los resultados de los estudios de coagulación de rutina se pueden utilizar para deducir los posibles trastornos (Fig. 1, Tabla 3). En forma aislada, el TP y el TTPA, respectivamente, evalúan la integridad de las vías extrínseca e intrínseca de la cascada de la coagulación. Juntos, el TP y el TTPA evalúan la integridad de la vía común final de la cascada de la coagulación. Del mismo modo, el TP y el TTPA están simultáneamente prolongados en condiciones que implican deficiencias que abarcan todas las 3 vías, tales como la DFCKD. Debido a que la generación de fibrina no se ve afectada en la deficiencia de factor XIII, el TP y el TTPA son normales en este TCR.
Se recomienda consultar con un hematólogo para ayudar en la interpretación de los resultados de laboratorio, facilitar aún más las pruebas definitivas, y para asegurarse de que los estudios de laboratorio confirmatorios son realizados correctamente. Si está disponible, se puede realizar un estudio de mezcla combinando plasma de banco normal y plasma del paciente en una proporción 1:1 para determinar la causa de un TP o un TTPA anormal (Fig. 1). En las deficiencias cuantitativas de factores tales como los TCRs, la adición de plasma normal sustituye al factor deficiente (s), corrigiendo de esta manera el TP o el TTPA anormal. A la inversa, la falta de una corrección parcial en un estudio de mezcla sugiere la presencia de un anticuerpo del factor de coagulación específico o no específico (inhibidor). Los inhibidores inespecíficos incluyen a los anticoagulantes lúpicos, que pueden producirse transitoriamente en los niños (por ejemplo, después de una enfermedad viral), son generalmente asintomáticos, y tienden a ser la causa más común de TTPA prolongado en niños sin sangrados. Los inhibidores de factores específicos son mucho menos comunes en los niños, incluso menos comunes que la mayoría de los TCRs. Por lo tanto, cuando un TTPA prolongado o un TTPA y un TP prolongados al mismo tiempo no corrigen en un estudio de mezcla, se debe primero brindar una mayor investigación para el anticoagulante lúpico. Esta investigación es primero completada demostrando la fosfolípido-dependencia del inhibidor, dado que los anticoagulantes lúpicos caen dentro de esta categoría. La corrección del TTPA o de otra prueba de cribado basada en fosfolípidos, el tiempo de dilución Russell de veneno de víbora, después de la adición de una fuente fosfolipídica (por ejemplo, plaquetas en el procedimiento de neutralización plaquetaria) confirma la presencia de un anticoagulante lúpico. Si la evaluación de un inhibidor de un factor de coagulación está indicada por motivos clínicos o la prueba del anticoagulante lúpico es negativa, los factores de la coagulación de interés pueden ser reducidos en base al estudio de la coagulación sin corregir (Fig. 1).
Los TCRs son en general confirmados por la reducción de la actividad del factor correspondiente (s) en un estudio de actividad funcional, que es el ensayo comúnmente utilizado cuando se solicita el nivel de un factor. Debido a que los estudios de la coagulación de rutina son normales en la deficiencia de FXIII, deben utilizarse pruebas de "screening" alternativas cuando existe la sospecha (o se quiere descartar) esta deficiencia. Actualmente, se recomienda un ensayo de actividad funcional del FXIII cuantitativa (liberación de amonio o incorporación de aminas) para el cribado en lugar de la prueba de solubilidad del coágulo utilizada tradicionalmente, que es sensible sólo a bajos niveles de FXIII (< 0,5% al 5%) y por lo tanto puede hacer que se pierdan las deficiencias más moderadas. La deficiencia de FXIII es confirmada luego por la cuantificación del antígeno FXIII-A2B2 por ensayo inmunoenzimático, seguido por la cuantificación de los antígenos de las subunidades A y B si el antígeno FXIII-A2B2 está reducido.
Existen varias consideraciones importantes en la evaluación de laboratorio de los TCRs en niños. En primer lugar, los valores de referencia para los recién nacidos a término y prematuros en los primeros 6 meses de vida (y más allá, en algunos casos) se diferencian de los de los adultos, particularmente para el TTPA y todos los niveles de factores de la coagulación excepto para el FVIII. Debido a que los niveles de muchos factores aumentan durante el primer mes de vida, las deficiencias leves o equívocas pueden necesitar ser confirmadas una vez que el niño es mayor o por evaluación de ambos padres.
Como siempre, varios factores pueden afectar artificialmente los resultados del estudio de la coagulación, incluyendo la contaminación de la muestra con heparina y la falla en la recolección de la cantidad especificada de sangre dentro del tubo de recolección (para una óptima relación plasma-citrato). Se recomienda la mezcla inmediata y suave de la muestra con anticoagulante para evitar la activación de la coagulación. Las muestras deben idealmente ser analizadas dentro de las 4 horas de la recolección o, alternativamente, congeladas inmediatamente para su futuro uso. Si originalmente son elaboradas después de la administración empírica de plasma fresco congelado (PFC) para tratar un sangrado activo, las pruebas de coagulación deben repetirse antes de interpretar los resultados y concluir si un paciente tiene un TCR. El tiempo para volver a controlar los estudios de coagulación para descartar TCRs (u otros trastornos de la coagulación) dependerá de las vidas medias de los factores de la coagulación potencialmente afectados (ej., FXI en una anomalía aislada del TTPA; FVII cuando sólo el TP es anormal; FI, FII, FV o FX cuando ambos TTPA y TP son anormales; y FXIII cuando ambos TTPA y TP son normales; Fig. 1, Tabla 4). Los niveles de los factores individuales pueden igualmente verse afectados por la administración previa de PFC y por lo tanto, puede que tengan que ser revisados de nuevo después de que ha transcurrido la cantidad adecuada de tiempo (de nuevo, en base a la vida media; Tabla 4). En general, los estudios de la coagulación relevantes o los niveles del factor deben ser comprobados de nuevo después de que hayan transcurrido 5 vidas medias de eliminación del factor deficiente (s) sospechado si el objetivo es hacer un diagnóstico de TCR después de la administración de PFC para tratar un episodio hemorrágico inicial. Idealmente, las muestras para las pruebas hematológicas se deben recoger en ≥ 2 tubos de tapa azul (que contienen citrato de sodio como anticoagulante) antes de la administración de PFC o cualquier otro producto de reemplazo de factores cuando la presencia de un trastorno hemorrágico es posible o sospechada.
Tabla 3. Hallazgos en los estudios de laboratorio iniciales en los TCRs
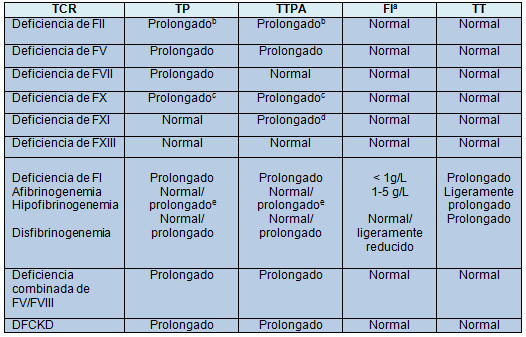
Los hallazgos excluyen los resultados del hemograma (el recuento de plaquetas será presumiblemente normal en todos los casos). TT, tiempo de trombina.
aRango normal = 1,5 – 3,5 g/L
bPuede estar solo mínimamente prolongado dependiendo de los reactivos utilizados: los resultados pueden estar también dentro del rango normal.
cSe han reportado variantes congénitas con TP y TTPA normales
dUn TTPA normal no excluye el diagnóstico de deficiencia de FXI leve, particularmente si los niveles de FVIII están elevados.
eEl TP o el TTPA pueden estar normales o prolongados en la hipofibrinogenemia, dependiendo de cuán bajo es el nivel de FI; el TP y el TTPA son típicamente normales cuando el nivel de FI excede 1g/L.
En situaciones en las que se sospecha un trauma no accidental, los médicos a menudo están justamente preocupados por descartar una diátesis hemorrágica subyacente. Aunque los hematomas subdurales en niños < 2 años de edad son principalmente causados por traumatismos no accidentales, particularmente si se asocian con hemorragias retinianas, hay informes anecdóticos de pacientes en los que esta combinación de hallazgos se atribuyó a una diátesis hemorrágica, incluyendo el sangrado por deficiencia de vitamina K en el recién nacido (también conocido como enfermedad hemorrágica tardía del recién nacido), la HA, la enfermedad de von Willebrand tipo 1 y el síndrome de Hermansky-Pudlak. Los trastornos hemorrágicos sólo rara vez han sido reportados como causa de hemorragias retinianas aisladas en neonatos. Múltiples fracturas de diferentes edades (especialmente fracturas costales posteriores en lactantes con hematoma subdural) o inconsistencias entre los mecanismos reportados y el patrón o el alcance de las lesiones sugieren un trauma no accidental, especialmente cuando hay retraso en la búsqueda de atención médica. Por el contrario, debe considerarse la posibilidad de TCRs en niños de padres consanguíneos o con una historia familiar o personal de sangrado, en particular en ausencia de otras características que sugieren trauma no accidental. Las recomendaciones para la consideración de los diversos trastornos hemorrágicos, incluyendo TCRs, en el entorno de un posible trauma no accidental se resumieron recientemente en un informe técnico de la Academia Americana de Pediatría. La presencia de un trastorno hemorrágico y de un trauma no accidental no es mutuamente excluyente, por lo que la posibilidad de que este último debe siempre tenerse en cuenta, sobre todo si se aplican algunas de las características antes mencionadas que sugieren trauma no accidental.
Tabla 4. Vidas medias de los factores de la coagulación

Manejo
En general, existe relativamente poca información con respecto al manejo de individuos con TCRs, aunque las guías para el manejo de los episodios de sangrado, cirugía y embarazo (incluyendo la atención del potencial recién nacido afectado) se han propuesto en base a la literatura existente y a la experiencia clínica. Sin embargo, dada la rareza de los TCRs y la naturaleza compleja y a menudo costosa del tratamiento, los niños afectados requieren ser seguidos en un centro de tratamiento para hemofilia al menos bianualmente para garantizar no solo el cuidado apropiado, sino también para facilitar el acceso a terapias que pueden no estar inmediatamente disponibles en cualquier lugar. El centro de tratamiento de la hemofilia debe ser consultado acerca del manejo de cualquier sangrado así como de la cobertura hemostática y las precauciones ante procedimientos invasivos. No todos los eventos hemorrágicos requieren tratamiento. En general, la necesidad de cobertura hemostática para cirugía u otros procedimientos invasivos se basa en el riesgo general de sangrado atribuible a la respectiva deficiencia del factor y al procedimiento en sí, y en los antecedentes personales y familiares de sangrado con los desafíos hemostáticos previos, incluyendo la cirugía.
Productos de Reemplazo
Los productos de reemplazo disponibles en los Estados Unidos para el tratamiento o la prevención de sangrados en los TCRs, cuando está indicado, se resumen en la Tabla 5.
Cuando se dispone de un concentrado de un solo factor específico, ese es el tratamiento de elección. En los Estados Unidos, los concentrados de factores únicos están disponibles para todos los factores deficientes abarcados por los TCRs excepto para el FII, el FV, el FX (concentrado en desarrollo clínico) y el FXI (concentrado disponible sólo en Europa). Para las deficiencias de FII y FX, se recomiendan los concentrados de complejo de protrombina (CCP). Los CCP son concentrados de factores de la coagulación específicos altamente purificados obtenidos de un pool de plasma normal, es decir, FII, FIX, FX, y FVII en cantidades variables dependiendo del producto. Los CCPs contienen cantidades conocidas de los factores involucrados y que son sometidos a un proceso de inactivación vírica, siendo preferibles al PFC y al crioprecipitado cuando el objetivo es sustituir el FII o el FX.
El PFC se utiliza a menudo como una fuente alternativa de reemplazo de factores en los TCRs y es uno de los pilares del tratamiento de la deficiencia de FV, en ausencia de cualquier otro producto que contiene este factor. El PFC también puede ser utilizado cuando se indica tratamiento hemostático o cobertura en la deficiencia de FXI en los Estados Unidos. El crioprecipitado, un subproducto del PFC obtenido después de la descongelación de un volumen de PFC de un solo donante a 4° C, es rico en factor de von Willebrand, FVIII, FXIII, y FI, y se utiliza con mayor frecuencia para el reemplazo del FI y del FXIII, aunque más recientemente, los productos purificados derivados del plasma han estado disponibles para tratar estas últimas 2 deficiencias. Aunque ampliamente disponibles y relativamente baratos, el PFC y el crioprecipitado tienen desventajas potenciales en comparación con los concentrados de factor único y CCPs en que no se someten a un proceso de inactivación viral en los Estados Unidos (aunque los donantes y el plasma son examinados exhaustivamente) y, dadas las cantidades relativamente pequeñas de los factores individuales en el mismo, son a menudo requeridos en grandes volúmenes para reemplazar un solo factor, lo que podría poner a los receptores en riesgo de sobrecarga de fluidos.
El tratamiento hemostático o la cobertura en la deficiencia combinada de FV/FVIII consisten en el reemplazo de ambos factores utilizando PFC para reemplazar el FV y los concentrados de FVIII derivados de plasma o recombinantes para reemplazar el FVIII (Tabla 5). Basado principalmente en la experiencia con warfarina, el PFC o el CCP se recomiendan para el tratamiento del sangrado o para la cobertura quirúrgica en la DFCKD, además del mantenimiento con vitamina K por vía oral.
Tabla 5. Productos de reemplazo de factores actualmente disponibles en los Estados Unidos para el tratamiento o prevención de sangrados en los TCRs.(Ver tabla)
Terapias auxiliares
Las terapias auxiliares que se pueden utilizar como alternativas o complementos al reemplazo de factores incluyen antifibrinolíticos sistémicos (es decir, ácido tranexámico y ε-ácido aminocaproico), agentes hemostáticos tópicos (por ejemplo, adhesivo de fibrina), y, en el ámbito de la deficiencia combinada de FV/FVIII, desmopresina para aumentar los niveles de FVIII. En algunos casos, los antifibrinolíticos pueden ser utilizados exclusivamente para el sangrado de la mucosa o para procedimientos menores (por ejemplo, dentales) evitando el uso de productos sanguíneos mancomunados. Para la menorragia relacionada con TCRs, las terapias hormonales y los antifibrinolíticos son a menudo terapias de primera línea eficaces y consideradas, como lo son en las mujeres sin trastornos hemorrágicos.
Terapia de reemplazo profiláctica
En algunos casos, puede estar justificada la terapia hemostática profiláctica, ya sea primaria (iniciada antes de que se produzca cualquier sangrado) o secundaria (iniciada después de un episodio de sangrado, para evitar la recurrencia). La profilaxis primaria ha sido utilizada con eficacia en niños con HA severa para reducir el daño articular por hemartrosis recurrente. Sin embargo, los datos con respecto a cualquier beneficio de la profilaxis primaria en los TCRs son escasos. El único TCR para el que la profilaxis está recomendada habitualmente es la deficiencia severa de FXIII, en la que se recomienda la profilaxis desde el momento del diagnóstico debido al riesgo de eventos hemorrágicos graves, e incluso potencialmente mortales, como HIC. La profilaxis secundaria puede ser considerada después del sangrado musculo-esquelético o de una hemorragia potencialmente mortal para prevenir el sangrado recurrente, particularmente en las deficiencias de factor XIII, FX, y FVII y en casos severos de deficiencia de FI y FV.
Recomendaciones perinatales
El parto de un recién nacido que puede resultar afectado debe realizarse idealmente en un centro especializado, con planificación anticipada para limitar el riesgo de hemorragia en la madre y el neonato. El monitoreo del cuero cabelludo fetal y el parto asistido con fórceps o vacío deben ser desalentados. La comunicación entre el centro de tratamiento de la hemofilia, el obstetra, y el pediatra o el neonatólogo es clave para evitar consecuencias graves, como HIC, en el recién nacido potencialmente afectado y para considerar el manejo a partir de entonces. Después del parto está indicada la evaluación diagnóstica completa del neonato; se puede enviar la sangre del cordón si hay antecedentes familiares conocidos de un TCR, dada la facilidad de recolección y para evitar el sangrado de tejidos blandos relacionado con la punción venosa. Los desafíos hemostáticos (por ejemplo, la circuncisión, las punciones arteriales) deben ser evitados hasta que se obtengan los resultados. Se recomienda realizar una ecografía craneal durante la primera semana de vida en los recién nacidos con TCRs graves conocidos para excluir la presencia de HIC.
Recomendaciones pediátricas adicionales
En el período neonatal y posteriormente, debe considerarse la posibilidad de administrar las medicaciones (incluyendo la vitamina K en el recién nacido) y las inmunizaciones por vía subcutánea (en lugar de por vía intramuscular), cuando sea posible. En particular, las vacunas contra la hepatitis A y B deben ser dadas en todos los niños como recomiendan los Centros para el Control y Prevención de Enfermedades. El cuidado dental preventivo de rutina debe ser alentado y facilitado, consultando con el centro de tratamiento de hemofilia sobre la necesidad de profilaxis y otras medidas hemostáticas. Los padres deben evitar dar a los niños con TCRs analgésicos/antipiréticos que pueden afectar la función plaquetaria (por ejemplo, antiinflamatorios no esteroides). Los profesionales y los padres pueden consultar los recursos existentes (por ejemplo, en la Fundación Nacional de Hemofilia y la Federación Mundial de Hemofilia, Sitio Web en www.wfh.org) y los centros de tratamiento de hemofilia para orientación relativa a la actividad física y otros aspectos de la atención de los niños y adolescentes con TCRs. Por último, se debe ofrecer consejo genético y evaluación a toda la familia para identificar potenciales portadores en riesgo de descendencia afectada, especialmente en países donde la incidencia de consanguinidad es alta o cuando una mutación genética específica ha sido identificada en la familia.
Conclusiones
La identificación temprana de los TCRs en niños es fundamental para garantizar un tratamiento óptimo de los eventos hemorrágicos y para mitigar cualquier riesgo de sangrado futuro. El sangrado puede ser grave, incluso mortal, y dada la naturaleza congénita de estos trastornos, los síntomas pueden comenzar durante el período neonatal o la infancia temprana. Los episodios hemorrágicos graves, incluyendo la HIC, pueden estar precedidos de eventos de sangrado relativamente menores, proporcionando una oportunidad para el diagnóstico antes de que ocurran. El pediatra y otros profesionales no hematólogos deben estar familiarizados con la presentación clínica variable y con la evaluación diagnóstica inicial de estos raros trastornos. Los TCRs deben ser considerados, en particular, cuando hay consanguinidad. La consulta con un hematólogo que está idealmente afiliado a un centro de tratamiento de hemofilia facilitará el diagnóstico eficaz y el manejo adecuado.
Comentario: La identificación de un niño con trastornos hemorrágicos puede resultar difícil, dada la frecuencia relativa de sangrado no patológico en las diversas edades y la presentación clínica variable de estos cuadros clínicos. La presencia de hemorragias menores o graves, el antecedente de consanguinidad y la historia familiar de sangrados deben hacer sospechar la presencia de un trastorno de la coagulación. La identificación precoz de los TCRs en niños ayudará al tratamiento de adecuado de los mismos, evitando sangrados recurrentes y mejorando la calidad de vida del paciente.
♦ Resumen y comentario objetivo: Dra. María Eugenia Noguerol