La queratodermis palmoplantar (PPK) pertenece a un grupo de enfermedades cutáneas hereditarias y adquiridas generalmente autosómicas dominantes y sus defectos genéticos se han identificado en algunos grupos. En éstos casos generalmente se reportan características extracutáneas. Debido a la rareza de las PPK hereditarias, los conocimientos se basan en reportes de casos. Una de las queratodermias heredadas es el síndrome de Vohwinkel (forma clásica y variante).
Presentación del caso:
Se presentan dos hermanos, uno masculino y uno femenino. La mujer afectada es la primera hija y el hombre afectado el último. Una hija es normal.
La mujer afectada, de 34 años, tiene pelo fino, escaso, seco, frágil y cuero cabelludo hipopigmentado, raramente excedía los 8 cm de longitud desde el nacimiento. El pelo de las cejas, pestañas y púbico, también era escaso. Al año de edad cuando comenzó a caminar, apareció una hiperqueratosis marginal de las palmas y plantas. Un claro borde con un halo inflamatorio rojizo en las superficies dorsales de las manos y pies estaba presente alrededor de la hiperqueratosis. Se desarrolló una banda constrictora alrededor de la articulación interfalángica distal del tercer dedo de la mano derecha a los 4 años. Las uñas, dientes y mucosas eran normales (figuras 1a, 1b, y 1d)
.jpg)
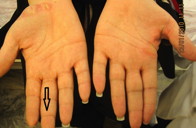
Figura 1a Figura 1b
Figura 1a. Hipotricosis del cuero cabelludo en la hermana.
Figura 1b. Queratodermia focal estriada en las palmas de la hermana, nótese la banda constrictora.
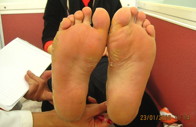
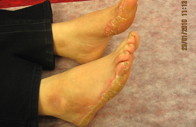
Figura 1c Figura 1d
Figura 1c. Queratodermia focal en las plantas de la hermana.
Figura 1d. Márgenes con halo eritematoso en la hermana.
El varón afectado, de 20 años, era aparentemente normal al nacer. El pelo cubría el cuero cabelludo durante los primeros 12 meses. A los 20 años se hizo escaso y fino como el de la hermana. El pelo de las cejas, pestañas, barba, bigote y púbico era escaso. Durante la infancia, se desarrolló una queratodermia focal (queratodermia estriada) en los puntos de presión en dedos de pies y talones bilateral. Se observaba un halo eritematoso alrededor de la hiperqueratosis de palmas y plantas. A los 5 años se desarrollaron bandas constrictoras alrededor de las articulaciones interfalángicas distales del segundo y tercer dedo de la mano derecha. Los dientes y mucosas eran normales. Los pacientes se quejaban de dolor en los dedos afectados causado por la congestión (figuras 2a, 2b, 2c y 2d). No tenían historia de sordera ni piel ictiósica y todas las características músculo esqueléticas eran normales. Los pacientes habían utilizado queratolíticos más emolientes con mejoría parcial y transitoria de la queratodermia. La historia familiar para enfermedad similar era negativa.
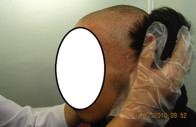

Figura 2a Figura 2b
Figura 2a. Hipotricosis del cuero cabelludo en el hermano.
Figura 2b. Banda constrictora del segundo y tercer dedo (nótese la congestión de los dedos).

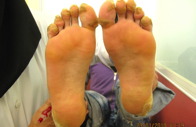
Figura 2c Figura 2d
Figura 2c. Márgenes con halo eritematoso en hermano.
Figura 2d. Queratodermia focal estriada de las plantas en el hermano.
Las investigaciones adicionales se detallan a continuación:
1. Las muestras de tejido obtenidas de piel plantar en ambos pacientes mostró las mismas características histopatológicas, hiperqueratosis densa con una membrana de paraqueratosis, hipergranulosis y acantosis regular de la epidermis (figura 3).
2. Los tricogramas del pelo del cuero cabelludo fueron iguales y excepto en la variabilidad de los diámetros del tallo piloso, los bulbos y las proporciones de catágeno, telógeno a anágeno eran normales (cerca del 10%).
3. Las audiometrías de los pacientes eran normales.
4. Se realizó el estudio genético con el propósito de detección de dos genes conocidos para el Síndrome de Vohwinkel (gen loricrina y conexina).
Los métodos y resultados del análisis genético se muestran abajo:
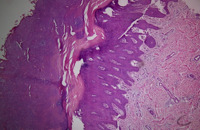
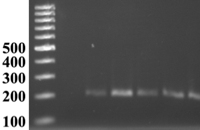
Figura 3. Figura 4.
Figura 3. Biopsia de la queratodermia plantar. Hiperqueratosis densa, paraqueratosis, acantosis regular y membrana granulosa preservada (H&E x40).
Figura 4. PCR amplificó un producto 201-bp, que contiene una parte de la región que codifica el gen loricrina: 1) padre, 2) madre, 3) hermano afectado, 4) hermana afectada, 5) hermano normal.
El ADN genómico se preparó de los pacientes, sus padres y el otro hermano. La secuencia directa del producto de PCR de la región que codifica el gen de loricrina no mostró la mutación en los pacientes presentados, sus padres ni hermano.
Otra investigación fue el análisis de secuencia directa del producto de PCR en la región que codifica el gen connexina 26, que mostró una mutación heterocigota silente 257-258GC-CG (código para el mismo aminoácido) en connexina 26 de los pacientes.
Se aconsejó a los pacientes que utilizaran queratolíticos con emolientes (urea al 10%). El paciente masculino que presentaba bandas constrictoras dolorosas alrededor del segundo y tercer dedo fue enviado a un cirujano de mano para la corrección quirúrgica.
Vohwinkel reportó una queratodermia mutilante y difusa palmoplantar en una mujer de 24 años en el año 1929. Desde entonces varios reportes han aclarado la clasificación, defectos genéticos y detalles clínicos de éste síndrome. Estos estudios incluyeron estudios genéticos.
Otros hallazgos reportados del síndrome de Vohwinkel clásico son sordera-mudez, alopecia Universalis congénita, alopecia tipo pseudopelada, acantosis nigricans, paraplejía espástica, miopatía, cambios ungueales, retardo mental, lesiones ampollares en las plantas y convulsiones.
Similar a los reportes de Arfan Bari y Mir Mubashir Ali, los pacientes presentados no mostraron las asociaciones comunes a las dos formas de síndrome de Vohwinkel (sordera e ictiosis).
Presentaban hipotricosis congénita, que según el conocimiento de los autores no se había reportado previamente. Los autores decidieron investigar las dos mutaciones conocidas en connexina 26 y loricrina en los pacientes estudiados. Sorprendentemente, el gen loricrina era normal y se detectó una mutación silente en el gen connexina 26. Debido a que los pacientes tenían hipotricosis difusa y congénita, también se deben considerar a los síndromes de displasia ectodérmica con PPK, especialmente el síndrome de Clouston, que se caracteriza por distrofia ungueal, hipotricosis difusa y PPK. La característica del síndrome de Clouston es la distrofia ungueal, no observada en éstos pacientes. El síndrome de Clouston tiene ortohiperqueratosis con una granulosa normal en la histopatología de PPK. En los pacientes estudiados se descarta el síndrome de Clouston porque presentaban uñas normales e hipergranulosis.
En conclusión, se reporta una queratodermia mutilante con 1) patrón estriado con borde rojizo, 2) hipotricosis congénita y 3) herencia autosómica recesiva en dos hermanos que puede considerarse como otro subgrupo del síndrome de Vohwinkel. Según el conocimiento de los autores no existen reportes similares. Se están realizando análisis genéticos en el futuro para encontrar la mutación responsable,
¿Qué aporta éste artículo a la práctica dermatológica?.
El Síndrome de Vohwinkel (queratodermis palmoplantar mutilante y difusa) se asocia con varias características incluyendo la ictiosis y la sordera. El modo de herencia es autosómica dominante con mutación en los genes loricrina y conexina.
Se reporta el caso de una queratodermia palmoplantar mutilante y focal en dos hermanos con hipotricosis congéntita y probable herencia autosómica recesiva que parece ser una nueva variante del síndrome de Vohwinkel.
Tabla 1. Especificación de los pacientes y de los dos subgrupos del síndrome de Vohwinkel.
| Modo de herencia | Locus del gen | PPK | Pseudoa inhum | Inicio | Queratosis extensora | Borde eritema toso | Caracterís ticas extracu táneas | Histopa tología | |
| Sd Vohwinkel (clásico) | AD | 13q11, q12 (connexin 26) | Difusa | + | Infancia | + | + | Sordera | Hiperqueratosis, paraqueratosis, acantosis e hipergranulosis |
| Sd Vohwinkel (variante) | AD | 1q21 (loricrin) | Difusa | + | Infancia | + | + | Ictiosis | Ictiosis Hiperquer atosis, paraqueratosis, acantosis e hipergranulosis |
| Pacientes estudiados | AR | ¿? | Focal | + | Infancia | - | + | Hiperqueratosis | Hiperqueratosis, paraqueratosis, acantosis e hipergranulosis |
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodriguez Rivello