Resumen
Las características morfológicas de la médula ósea son el pilar del diagnóstico en la trombocitemia esencial (TE) y la policitemia vera (PV). Las mutaciones descubiertas recientemente en JAK2, CALR y LMP son útiles para el diagnóstico. Estos pacientes tienen una mutación en JAK2 y su ausencia hace que el diagnóstico sea poco probable. La mayoría de los pacientes sin mutación en JAK2 expresan mutaciones en CALR o LMP.
Se considera que los pacientes <60 años sin antecedentes de trombosis son de bajo riesgo. El tratamiento en la PV de bajo riesgo es la flebotomía para lograr un hematocrito de 45% y la aspirina en dosis bajas. El tratamiento en la TE es la observación si no hay factores de riesgo para trombosis arterial o la aspirina en dosis bajas, una o dos veces por día, según los factores de riesgo. El tratamiento citorreductor se indica en pacientes de alto riesgo (>60 años o antecedentes de trombosis) en forma de hidroxiurea como primera línea e interferón alfa o busulfán como segunda línea.
Abreviaturas
LMA Leucemia mieloide aguda
LMP Leucemia mieloproliferativa
MFP Mielofibrosis primaria
MO Médula ósea
NMP Neoplasias mieloproliferativas
PV Policitemia vera
SMD Síndrome mielodisplásico
TE Trombocitemia esencial
TR Trombocitosis reactiva
Introducción
La trombocitemia esencial (TE) y la policitemia vera (PV) son neoplasias mieloproliferativas (NMP) caracterizadas por trombocitosis y eritrocitosis clonales, respectivamente. La mediana de edad al diagnóstico es de 71 años para ambas. La preponderancia es femenina para la TE y masculina para la PV.
Los pacientes con TE o PV también pueden tener leucocitosis, esplenomegalia y síntomas microvasculares, entre ellos cefaleas, trastornos visuales (ej, visión borrosa), mareos, palpitaciones, dolor torácico atípico, parestesias, eritromelalgia (eritema, calor y dolor en las extremidades distales), prurito (en general después de bañarse) y otros síntomas constitucionales, como cansancio.
La evolución clínica de ambas enfermedades se puede interrumpir por complicaciones trombohemorrágicas y transformación de la enfermedad en mielofibrosis (MF) o leucemia mieloide aguda (LMA). La expectativa de vida en pacientes con TE es inferior a la de controles emparejados para edad y sexo, con una mediana de supervivencia estimada en 20 años para la TE y 14 años para la PV; en pacientes más jóvenes, las medianas de supervivencia fueron 33 y 24 años. Las tasas de transformación leucémica o fibrótica a 20 años se estiman en el 10% - 20% para la PV y el 5% - 10% para la TE.
Los pacientes con PV o TE se caracterizan por la presencia de mutaciones somáticas específicas para NMP, como Janus cinasa 2 (JAK2), la calreticulina (CALR), y el oncogen del virus de la leucemia mieloproliferativa (LMP). Se cree que estas tres mutaciones son patogenéticamente esenciales y a menudo se producen de manera mutuamente exclusiva. La PV casi siempre se asocia con una mutación de JAK2. (98% JAK2 V617F).
En la TE, las frecuencias de mutaciones en JAK2V617F y CALR son de aproximadamente el 60% y el 22%, respectivamente. Se están estudiando la incidencia y la importancia de otras mutaciones no específicas que podrían coexistir con las ya mencionadas específicas para NMP.
Asimismo, se hallaron mutaciones en JAK2 en otras neoplasias mieloides malignas, aunque con mucha menor frecuencia; siendo una excepción la enfermedad de superposición SMD/NMP, llamada anemia refractaria con sideroblastos en anillo asociados con importante trombocitosis (RARS-T), en la que la frecuencia de mutaciones es de hasta el 50%.En general, los pacientes con mutaciones en JAK2 con TE son de más edad que los que tienen mutaciones en CALR y tienen valores más altos de hemoglobina y leucocitos y valores menores de plaquetas, así como mayor riesgo de trombosis.
Diagnóstico
Además de la TE y la PV, la clasificación de la Organización Mundial de la Salud (OMS) incluye otra seis entidades dentro de las NPM y emplea las características morfológicas de la médula ósea (MO) (ahora reemplazadas por los estudios genéticos) para distinguir las NMP del SMD y las enfermedades de superposición SMD/NMP.
Un proceso similar de diagnóstico morfológico complementado por el análisis de la mutación se aplicó para formular los criterios diagnósticos basados sobre la OMS para la TE y la PV, que están siendo revisados según una propuesta reciente. Sin embargo, en la práctica clínica habitual es deseable un enfoque más práctico, que se verá a continuación.
Policitemia Vera La policitemia vera se sospecha cuando se comprueba el aumento de los valores del hematocrito o la hemoglobina. Este aumento puede ser real (asociado con aumento de la masa de eritrocitos) o aparente (no asociado con aumento de la masa de eritrocitos). Otros motivos para sospechar PV son el prurito causado por el agua y la trombosis de una gran vena abdominal (por ejemplo, la vena hepática o la vena porta). Además de la PV, el diagnóstico diferencial del aumento de la hemoglobina/hematocrito incluye la eritrocitosis congénita y adquirida.
El primer paso en la evaluación de estos pacientes es la pesquisa de mutación de JAK2V617F; este primer paso diagnóstico identifica alrededor del 95% de los pacientes con PV. También se recomienda medir la eritropoyetina (Epo) porque los valores subnormales confirman el diagnóstico si está presente la mutación en JAK2V617F, pero también sugieren PV con JAK2V617F no mutado en ausencia de la mutación.
Este último caso justifica la pesquisa de otras mutaciones en JAK2 cuya presencia avala el diagnóstico de PV y en su ausencia es necesario el examen de MO para descartar una PV sin mutación de JAK2, que podría ser el caso en el 1% de los pacientes.
El examen de la médula ósea es especialmente útil para distinguir la TE con mutación en JAK2 de la PV “enmascarada”. En este caso el paciente tendrá características morfológicas medulares coincidentes con PV (es decir hiperplasia de las tres líneas celulares con desviación a la izquierda e hiperplasia megacariocítica y pleomórfica con características atípicas), pero no tiene el valor umbral de hemoglobina exigido por los criterios de 2008 de la OMS (>18,5 g/dl en hombres y >16,5 g/dl en mujeres).
Esa situación es uno de los fundamentos de la propuesta de una nueva revisión de los criterios diagnósticos de la OMS para PV, en la que se agregaron las características morfológicas de la MO como un criterio diagnóstico principal y se bajó el umbral de hemoglobina a 16,5 g/dl para os hombres y 16 g/dl para las mujeres.
La ausencia de mutación en JAK2 junto con un nivel plasmático normal o aumentado de Epo hacen que el diagnóstico de PV sea improbable y justifica investigar la eritrocitosis congénita o adquirida. Se recomienda la revisión de los registros anteriores de hemogramas completos para determinar si el aumento de los valores de hemoglobina/hematocrito es nuevo o crónico. Este último sugiere eritrocitosis congénita y el primero trastornos adquiridos que suelen producir hipoxia central o periférica.
El diagnóstico diferencial de eritrocitosis adquirida que no es PV incluye procesos causados por hipoxia y procesos independientes de la hipoxia. Entre los primeros se encuentran el hábitat a grandes alturas, la enfermedad pulmonar hipóxica, los shunts cardiopulmonares de derecha a izquierda, el tabaquismo intenso, la intoxicación por monóxido de carbono, la apnea del sueño y la estenosis de la arteria renal.
Las causas de eritrocitosis adquirida independiente de la hipoxia son algunos fármacos (por ejemplo estimulantes de la eritropoyesis, preparados con testosterona), tumores productores de Epo (carcinoma de células renales, carcinoma hepatocelular, hemangioblastoma cerebeloso, meningioma, feocromocitoma, leiomioma uterino, adenoma paratiroideo), quistes renales y trasplante renal.
Trombocitemia esencial
La trombocitosis es frecuente y en la mayoría de los casos representa un proceso reactivo.
Las causas de la trombocitosis reactiva (TR) son las infecciones, el daño tisular, las neoplasias malignas, la inflamación crónica, la hemólisis, ciertos fármacos, la anemia ferropénica y la esplenectomía.
La trombocitosis no reactiva adquirida a menudo indica un trastorno mielode maligno, entre ellos la TE. La trombocitosis congénita es muy rara.
Debido a que la TR es la causa más frecuente de trombocitosis hallada en la práctica médica, es importante usar el juicio clínico antes de comenzar con el análisis de mutaciones como primer paso de la evaluación. Los marcadores clonales empleados para una trombocitosis inexplicada y persistente son JAK2V617F (presente en alrededor del 60% de los pacientes con TE, mielofibrosis primaria [MFP] , o RARS-T y el 95% de los pacientes con PV), las mutaciones en CALR (presentes en el 20%-25% de pacientes con TE o MFP), mutaciones en LMP (en el 3%-7% de los pacientes con TE o MFP), BCR-ABL1 (en el 100% de los pacientes con leucemia mieloide crónica) y mutaciones en SF3B1 (en aproximadamente el 80% de los pacientes con RARS-T). Se recomienda comenzar con la pesquisa de JAK2V617F y BCR-ABL1. i los resultados son negativos, seguir con CALR y, si éste es negativo efectuar la pesquisa de LMP.
Se recomienda también el examen de la MO para evaluar la trombocitosis inexplicada, ya que la ausencia de mutaciones no descarta la posibilidad de TE (el 15% de los pacientes pueden ser de tipo salvaje para JAK2, CALR, o LMP- llamados casos triple negativos), y la presencia de JAK2, CALR o LMP no puede de por sí distinguir entre TE y MFP.
La médula ósea en pacientes con TE o NMP relacionada, a menudo muestra hipercelularidad y formación de agrupación de megacariocitos, ambos ausentes en la TR. Asimismo, la proliferación megacariocítica en la TE se asocia con características morfológicas grandes y maduras con formación de conglomerados sueltos, mientras que en la MFP se asocia con índice núcleocitoplasmático aberrante y núcleos hipercromáticos y plegados irregularmente, agrupamiento denso, proliferación granulocítica y disminución de la eritropoyesis. Además, la reticulina o la fibrosis del colágeno podrían no ser fácilmente aparentes en la MFP prefibrótica.
Pronóstico
Supervivencia
Un estudio reciente de la Mayo Clinic y de Italia con 1581 pacientes incluyó 826 pacientes de la Mayo Clinic, 292 con TE y 267 con PV. Al 58% de estos dos últimos grupos se les efectuó seguimiento hasta su muerte. La mediana de supervivencia fue significativamente mayor en la TE que en la PV (19,8 años y 13,5 años, respectivamente). La mediana de supervivencia para pacientes menores de 60 años fue de 32,7 años para la TE y 23,8 años para la PV. La expectativa de vida para los pacientes con TE fue inferior a la de la población de los EEUU emparejada para edad y sexo (P<=0,001). La ventaja de supervivencia de la TE sobre la PV no fue afectada por la edad, el sexo o las mutaciones (es decir, JAK2 vs CALR vs LMP mutados vs triple-negativo).
Transformación leucémica, fibrótica y policitémica
En la cohorte de la Mayo Clinic del estudio mencionado, se informó la transformación leucémica en 18 de los 267 pacientes con PV (6,7%) y en 12 de los 292 pacientes con TE (4,1%) y la transformación fibrótica en 34 pacientes con PV (12,7%) y en 29 pacientes con TE (9,9%).
En la cohorte italiana de 594 pacientes (310 con PV y 284 con TE), se informaron 91 transformaciones fibróticas, incluidos 65 en pacientes con PV (21,0%) y 26 en pacientes con TE (9,2%) (P<0,01). La conversión de TE a PV fue infrecuente tanto en la cohorte de la Mayo Clinic (3%) como en la italiana (2,1%).
Factores de riesgo para la supervivencia, la transformación leucémica, y la progresión fibrótica
Los factores de riesgo son similares para la TE y la PV: edad avanzada, leucocitosis y antecedentes de trombosis.
Además un cariotipo anormal se asoció con peor supervivencia en la PV, independientemente de los factores de riesgo antes mencionados. En un estudio de PV, la edad >70 años, el recuento de leucocitos <13X 109/l y la ausencia de trombosis se asociaron con supervivencia relativa a 10 años del 84% vs el 59% en presencia de 1 y del 26% en presencia de 2 o más de estos factores de riesgo.
En la TE, la edad < 60 años, los valores normales de hemoglobina y el recuento de leucocitos <15 X 109/l se asociaron con una mediana de supervivencia de más de 20 años vs 9 años en presencia de 2 o más de estos factores de riesgo. Los factores de riesgo para la transformación leucémica de la PV son la ancianidad, la leucocitosis y el cariotipo anormal. Los factores de riesgo para la transformación fibrótica son una carga de alelo JAK2V617F mayor del 50%.
En una población de estudio de pacientes con TE y MFP prefibrótico, la supervivencia fue significativamente menor en presencia de características morfológicas de MFP prefibrótico; edad avanzada; antecedentes de trombosis; leucocitosis y anemia. La supervivencia sin leucemia también fue afectada adversamente por las características morfológicas de la MFP prefibrótica y los antecedentes de trombosis, pero también por la trombocitosis extrema. Asimismo, las características morfológicas de la MFP prefibrótica, la ancianidad, la anemia y la ausencia de mutación de JAK2 se asociaron con aumento del riesgo de progresión fibrótica.
Factores de riesgo de trombosis
Estudios recientes señalaron la diferencia en la frecuencia y el perfil de factor de riesgo de la trombosis arterial vs la trombosis venosa en la PV y la TE. Es bien sabido que el principal factor de riesgo de trombosis arterial y venosa es el antecedente de estos episodios.
Otros factores de riesgo para la trombosis arterial en la PV son el antecedente de hipertensión y para la trombosis venosa, la edad avanzada. Otros factores de riesgo para la trombosis arterial en la TE son edad<60 años, presencia de factores de riesgo cardiovascular (CV), leucocitosis y JAK2V617F y para la trombosis venosa, el sexo masculino. Es importante observar que es la ausencia de mutación de JAK2V617F y no necesariamente la presencia de mutación CALR o de estado mutacional triple negativo lo que se asocia con menor riesgo de trombosis en relación con los casos de mutación de JAK2.
Tratamiento
El tratamiento farmacológico actual para la PV o la TE no es curativo y no prolonga la supervivencia ni previene la transformación de la enfermedad en LMA o MF pos PV o pos TE. Se debe informar sobre esto a los pacientes. Es necesario que comprendan que el objetivo del tratamiento farmacológico es prevenir la trombosis en los pacientes de alto riesgo o aliviar los síntomas, si los hay.
Por otra parte, la mediana de supervivencia en la PV antes de la era de la flebotomía era de menos de 2 años, y la cifra actual de aproximadamente 14 años es una mejoría evidente. En la actualidad la flebotomía se considera el pilar del tratamiento de la PV y su valor se confirma en un estudio aleatorizado reciente en el que el objetivo de un hematocrito de 45% fue superior al del 45% - 50% para prevenir la trombosis recurrente.
El tratamiento farmacológico con antiplaquetarios (por ejemplo aspirina) o agentes citorreductores (por ejemplo hidroxiurea) se emplea en la PV o la TE para la profilaxis de la trombosis y el alivio sintomático. Estudios controlados hallaron reducción significativa del riesgo de trombosis con aspirina una vez al día en dosis bajas en la PV, tratamiento citorreductor con clorambucil o radiofósforo en la PV, tratamiento con hidroxurea en la TE de alto riesgo.
Sin embargo el empleo de clorambucil y radiofósforo causó menor supervivencia debido a la mayor generación de leucemia con estos fármacos. Otros mielosupresores, como el pipobroman y el anagrelide, también causaron complicaciones graves, como leucemia y mielofibrosis, respectivamente. El único fármaco con suficiente evidencia de eficacia y seguridad es la hidroxiurea, que es de primera elección en la PV o la TE de alto riesgo.
En casos resistentes a la hidroxiurea, datos de estudios no controlados apoyan el empleo de interferón alfa o busulfán como fármaco de segunda elección. El ruxolitinib (inhibidor de JAK) fue aprobado recientemente para tratar la PV intolerante o resistente a la hidroxiurea, pero genera inmunosupresión y aumenta el riesgo de infecciones oportunistas en pacientes con MF. Por lo tanto, habitualmente sólo se emplea el ruxolitinib para la PV en casos de esplenomegalia sintomática resistente a hidroxiurea, interferón alfa o busulfan o en casos de prurito intratable.
El “alto riesgo de trombosis” es la principal indicación de tratamiento citorreductor tanto en la PV como en la TE. Es importante por lo tanto precisar con exactitud la enfermedad de alto riesgo y limitar la quimioterapia a esos pacientes. Los antecedentes de trombosis y la edad avanzada (≥60 años) se identificaron como los factores de riesgo más importantes de trombosis, tanto en la PV como en la TE y su ausencia o presencia se emplean para estratificar a los pacientes en categorías de bajo riesgo y de alto riesgo, respectivamente. Estudios más recientes confirmaron el valor pronóstico independiente de los antecedentes de trombosis y la edad de 60 años o más para pronosticar los episodios trombóticos, así como la trombosis arterial y venosa consideradas por separado.
Además de los antecedentes de trombosis y la edad avanzada, estudios recientes también revelaron el valor pronóstico de las mutaciones en JAK2 y los factores de riesgo CV de trombosis arterial. El estado mutacional es especialmente importante en la TE, donde la presencia de JAK2V617F se asoció con mayor riesgo de trombosis que las mutaciones en CALR o el estado mutacional triple negativo. La asociación independiente entre JAK2V617F o factores de riesgo CV y trombosis también se destacó en otro gran estudio multicéntrico.
Los autores repasaron la estratificación de riesgo actual en TE y PV a la luz de esta asociación.(Figura) Los pacientes antes considerados de bajo riesgo (<60 años y sin antecedentes de trombosis) ahora se pueden subclasificar como “de muy bajo riesgo” y “de bajo riesgo” según la ausencia o la presencia de JAK2V617F o de factores de riesgo CV, respectivamente.(Figura).
El algoritmo terapéutico presentado en este artículo tiene en cuenta estas nuevas subclasificaciones y proporciona recomendaciones para reducir al mínimo los episodios trombóticos recurrentes y la administración innecesaria de medicamentos (Figura).
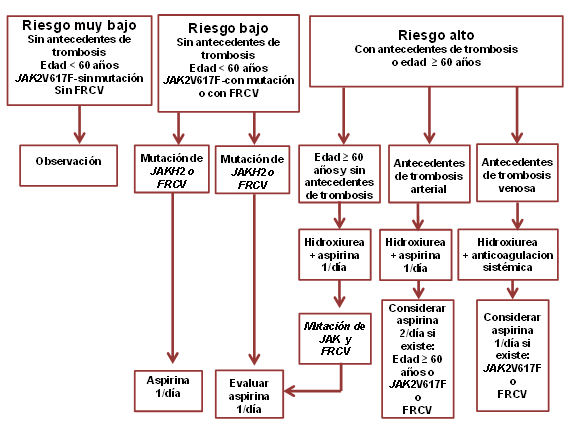
FIGURA. Algoritmo de tratamiento de la policitemia vera y la trombocitemia esencial. La aspirina se emplea en ausencia de contraindicaciones, incluido el síndrome de von Willebrand adquirido. Se recomienda la determinación de la actividad del cofactor de ristocetina en pacientes con más de 1 millón de plaquetas por microlitro y no indicar aspirina si el nivel de actividad es inferior al 20%. FRCV: factores de riesgo CV
En pacientes con trombosis venosa se recomienda la anticoagulación sistémica además del tratamiento citorreductor y su duración depende si la MP subyacente estaba bien controlada cuando se produjo el episodio trombótico. Si la enfermedad estaba bien controlada, se recomienda la anticoagulación sistémica por tiempo indefinido.
Si, por el contrario, la tromobosis se produjo en un paciente cuya enfermedad no estaba tratada de manera óptima y su hematocrito era superior al 45% o su recuento de plaquetas mayor de 450 X 109/l, es razonable indicar la anticoagulación sistémica por 6 meses, siempre que se hayan instituido otras medidas terapéuticas para controlar la enfermedad adecuadamente.
Discusión
Actualmente la pesquisa de mutaciones es parte integral del proceso diagnóstico de la PV y la TE. En ciertos casos, la detección de una mutación relativamente específica con un cuadro clínico evidente puede tornar innecesario el examen de la MO.
Asimismo, éste podría no ser esencial en pacientes ancianos para distinguir con exactitud la TE definida por la OMS de la mielofibrosis prefibrótica, ya que no afectaría demasiado el tratamiento específico. En todos los demás casos se recomienda el examen de MO para el diagnóstico exacto, el pronóstico y el tratamiento apropiados de la enfermedad.
El nuevo algoritmo (Figura) se basa sobre el hecho que el tratamiento actual para prevenir la trombosis es insatisfactorio así como sobre la nueva información acerca de la influencia significativa del estado mutacional y de los factores de riesgo CV sobre el riesgo de trombosis recurrente.
Los autores consideran que la flebotomía y el tratamiento con aspirina son las dos modalidades terapéuticas más importantes. Se basan en los resultados de estudios controlados y no controlados. Además, observaciones clínicas preliminares y estudios de laboratorio controlados revelaron el valor del tratamiento con aspirina dos veces al día en lugar de una vez y el posible valor de la aspirina para prevenir la trombosis venosa recidivante.
La flebotomía se indica en todos los pacientes con PV en quienes se desea que el hematocrito sea de 45%.
Con respecto al tratamiento farmacológico, quizás los pacientes “de muy bajo riesgo” con TE no necesiten ningún tratamiento, ni siquiera aspirina. En cambio, los pacientes “de bajo riesgo” con TE o PV necesitan aspirina (dosis de 81 mg), ya sea una o 2 veces al día, según la presencia respectiva de 1 o 2 o más de los factores mencionados recientemente para la trombosis arterial (JAK2V617F, factores de riesgo CV y edad >60 años).
En la enfermedad de alto riesgo, se emplea universalmente tratamiento citorreductor en pacientes con antecedentes de trombosis venosa o arterial. La hidroxiurea es el fármaco de primera línea y el interferón alfa o el busulfán los de segunda elección.
También se recomienda agregar aspirina una vez al día, con excepción de los pacientes que están recibiendo anticoagulantes sistémicos y no tienen factores de riesgo para trombosis arterial, excepto la edad avanzada. Además, la aspirina dos veces al día podría ser necesaria en pacientes de alto riesgo con antecedentes de trombosis arterial y un factor de riesgo más de trombosis arterial.
Otros aspectos del tratamiento de la PV y la TE son el síndrome de von Willebrand adquirido, que puede acompañar la trombocitosis extrema y así necesitar pesquisa de laboratorio para la actividad del cofactor de ristocetina; se debe evitar la aspirina cuando la actividad es menor del 20%.
El tratamiento para el prurito asociado con la PV comprende antihistamínicos, inhibidores selectivos de la recaptación de serotonina, inhibidores de JAK, interferón alfa, y fototerapia ultravioleta B de banda angosta. El tratamiento durante el embarazo incluye aspirina para las pacientes de bajo riesgo e interferón alfa para las de alto riesgo.
Conclusión
A pesar de varios descubrimientos importantes sobre las mutaciones en la TE y la PV, su patogénesis molecular aún no se conoce por completo. Es esencial tener mejor información a fin de crear fármacos que puedan modificar la evolución natural de estas enfermedades. Son necesarios estudios prospectivos para aclarar la importancia del tratamiento con aspirina dos veces al día y si el tratamiento citorreductor es verdaderamente esencial para los pacientes ancianos que no tienen mutación JAK2 ni antecedentes de trombosis.
Comentario y resumen: Dr. Ricardo Ferreira