La glomerulonefritis membranoproliferativa (GNMP), también denominada glomerulonefritis mesangial, se diagnostica sobre la base de un patrón de lesión glomerular que es común a un grupo heterogéneo de enfermedades. La GNMP representa aproximadamente el 7 al 10% de los casos de glomerulonefritis confirmada por biopsia y se ubica como la tercera o cuarta causa más importante de enfermedad renal terminal entre las glomerulonefritides primarias. A pesar de que algunas enfermedades asociadas a la GNMP son bien conocidas, los avances recientes han identificado otras condiciones asociadas a la GNMP
Presentación clínica
La presentación más común de la GNMP es en la infancia pero puede ocurrir a cualquier edad. La presentación clínica y la evolución son muy variables—de benigna y lentamente progresiva a rápidamente progresiva. Así, los pacientes pueden presentarse con hematuria asintomática y proteinuria, síndrome nefrítico agudo, síndrome nefrótico, insuficiencia renal crónica o, incluso, una glomerulonefritis rápidamente progresiva. La presentación clínica variada está causada por las diferencias en la patogénesis de la enfermedad y en el momento del diagnóstico dado mediante la biopsia en relación con la evolución clínica. El grado de insuficiencia renal también varía, y la hipertensión puede o no estar presente. Los pacientes que se presentan en la etapa inicial de la enfermedad con lesiones proliferativas en la muestra de la biopsia renal tienen más probabilidades de tener un fenotipo nefrítico; en cambio, aquellos con GNMP y semilunas pueden presentar una glomerulonefritis rápidamente progresiva. En contraste, los pacientes que en la muestra de biopsia muestran cambios avanzados que incluyen tanto la reparación como la esclerosis son más propensos a tener un fenotipo nefrótico. Los pacientes con GNMP clásica suelen tener características tanto de síndrome nefrítico agudo como de síndrome nefrótico—denominado síndrome de fenotipo nefrítico-nefrótico.
Clasificación y fisiopatología
Las características típicas de la GNMP en el microscopio de luz incluyen la hipercelularidad mesangial, la proliferación endocapilar y la remodelación de la pared capilar (con formación de dobles contornos)—todos los cuales dan como resultado la acentuación lobular de los ovillos glomerulares.
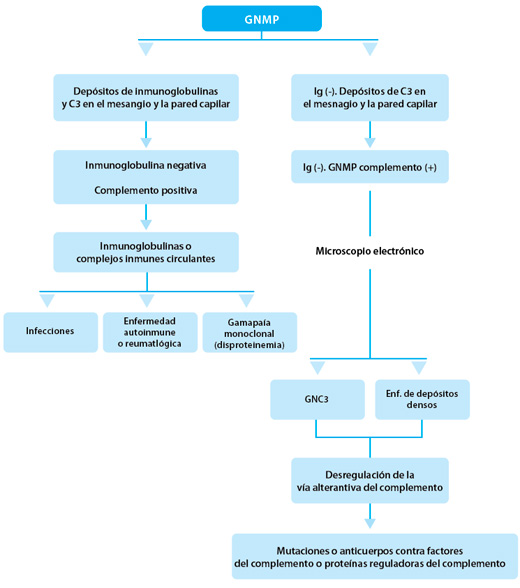
Estos cambios se deben a la deposición de inmunoglobulinas, factores del complemento, o ambos en el mesangio glomerular y a lo largo de las paredes capilares glomerulares. Con el microscopio electrónico, la GNMP tradicionalmente se clasifica como GNMP primaria tipo I (idiopática) (GNMP I), tipo II (GNMP II), o tipo III (GNMP III) o, GNMP secundaria. La GNMP I, la forma más común, se caracteriza por depósitos subendoteliales, y la GNMP III tiene depósitos tanto subepiteliales como subendoteliales. La GNMP II se caracteriza por depósitos densos en la membrana basal glomerular ("enfermedad de depósitos densos"). La GNMP secundaria, descrita por Rennke, es más frecuente debido a la hepatitis C y otras infecciones. Tal como están clasificadas, la GNMP I y la GNMP III probablemente incluyan tanto los casos de GNMP mediada por complejos inmunes como la GNMP mediada por el complemento.
Teniendo en cuenta los recientes avances en la comprensión del papel que tiene la vía alternativa del complemento en la GNMP, un enfoque práctico es considerar a la GNMP como mediada por complejos inmunes o mediada por el complemento. Por lo tanto, la GNMP mediada por complejos inmunes puede ocurrir cuando hay un aumento de los niveles circulantes de complejos inmunes mientras que la GNMP mediada por el complemento puede producirse debido a trastornos asociados a la desregulación de la vía alternativa del complemento.
GNMP mediada por complejos inmunes
Esta forma de GNMP es el resultado del depósito de complejos inmunes en los glomérulos debido a la antigenemia persistente, con complejos antígeno-anticuerpo que se forman a partir de infecciones crónicas, o de los niveles elevados de complejos inmunes circulantes secundarios a enfermedades autoinmunes, o debido a la paraproteinemia de las gammapatías monoclonales. Los complejos inmunes gatillan la activación de la vía clásica del complemento y la deposición de los factores del complemento de la vía clásica y la vía terminal del complemento, en el mesangio ya lo largo de las paredes capilares. El hallazgo clásico en la muestra de biopsia renal observada con microscopio de inmunofluorescencia es la presencia de inmunoglobulinas y complemento.
Hepatitis C y otras infecciones
Las infecciones crónicas virales como la hepatitis C y la hepatitis B, con o sin crioglobulinas circulantes, son una causa importante de GNMP. La hepatitis C, que fue reconocida como una causa común de GNMP mediada por complejos inmunes en la década de 1990, se considera ahora como la principal infección viral causante de GNMP. Además de las infecciones virales, las infecciones bacterianas crónicas (por ej., la endocarditis, la nefritis por derivación y los abscesos), las micosis y las infecciones parasitarias están asociadas a la GNMP, en particular en el mundo en desarrollo. Las bacterias asociadas a la GNMP incluyen al estafilococo, Mycobacterium tuberculosis, el estreptococo, Propionibacterium acnes, Mycoplasma pneumoniae, Brucella, Coxiella burnetii, nocardia, y el meningococo.
Enfermedades autoinmunes
La GNMP se produce en un número de enfermedades autoinmunes, incluyendo al lupus eritematoso sistémico y, en ocasiones, el síndrome de Sjögren, la artritis reumatoidea y la enfermedad mixta del tejido conectivo.
Gammapatía monoclonal
Estudios recientes indican que la deposición glomerular de la inmunoglobulina monoclonal como resultado de gammapatía monoclonal (también llamada disproteinemia o discrasia de células plasmáticas), con o sin crioglobulinas, se asocia con la GNMP. La gammapatía monoclonal se caracteriza por la proliferación de un solo clon de inmunoglobulinas producidas por los linfocitos o las células plasmáticas, resultando en la circulación de inmunoglobulina monoclonal. En un solo centro de estudio, el 41% de los pacientes con GNMP sin proceso autoinmune o infección crónica tenía evidencia de gammapatía monoclonal, según lo evaluado por medio de la electroforesis sérica, la electroforesis de orina, o ambas.
Las biopsias de la médula ósea en estos pacientes revelaron una variedad de condiciones: gammapatía monoclonal de significado incierto (GMSI) (la condición más común), linfoma de células B de bajo grado, linfoma linfoplasmacítico, leucemia linfocítica crónica y mieloma múltiple. Los autores sugieren que los pacientes con GNMP y gammapatía monoclonal deben ser clasificados como "gammapatía monoclonal asociada a GNMP" en lugar de GMSI.
GNMP mediada por el complemento
La cascada del complemento desempeña un papel importante en la inmunidad innata. Los factores del complemento pueden inducir la respuesta inflamatoria potente que da como resultado la quimiotaxis de los fagocitos, con la opsonización y la lisis de las células, incluyendo los microorganismos. La activación del complemento se produce a través de la vía clásica, la lectina o las vías alternativas, todas las cuales convergen para formar la C3 convertasa, que rompe C3 y lo convierte en C3a y C3b. En presencia de los factores B y D, C3b, se asocia con la C3 convertasa, generando incluso más C3 convertasa que resulta en un bucle de amplificación potente. Así, la C3 convertasa es un punto nodal en la cascada del complemento. La asociación de C3b y C3 convertasa también resulta en la formación de C5 convertasa, que activa la vía del complejo del complemento terminal y la formación del complejo de ataque a la membrana (C5b-C9) sobre la superficie celular, lo que provoca la lisis. La vía alternativa tiene continuamente un nivel de actividad bajo en la circulación (fase fluida) debido a la hidrólisis espontánea del enlace tioéster de C3 (mecanismo"a ralentí") que genera C3b; luego C3b se une a las membranas celulares y extracelulares tales como la membrana basal glomerular (fase superficial), así como a las membranas de microorganismos patógenos. Para evitar el auto-daño se produce la activación de la vía alternativa en un modo secuencial fuertemente regulado. En diferentes niveles de la cascada operan múltiples proteínas inhibidoras y reguladoras del complemento, en particular en el nivel de la C3 y la C5 convertasa. Dicho plasma o los reguladores de la fase fluida incluyen los factores H e I y la proteína 1 relacionada con el factor H a través de las proteínas reguladoras 5 y reguladoras ligadas a las células y en la superficie, como el factor de aceleración de la decadencia del complemento (CD55), el receptor del complemento 1, CD59, la proteína cofactor de membrana (CD46), y el receptor del complemento de la superfamilia de inmunoglobulinas.
El factor H acelera la ruptura de la C3 convertasa y es un cofactor para el clivaje mediado por el factor I y la inactivación de C3b, controlando de este modo la vía alternativa en la fase fluida. Los reguladores de la fase fluida del complejo del complemento terminal incluyen la vitronectina y la clusterina. Algunos de los reguladores de la fase líquida, incluyendo el factor H y los factores relacionados con la proteína H 1 también se adhieren a la superficie celular y a las membranas extracelulares, añadiendo un mecanismo de protección extra para evitar la formación de reguladores activos de los productos activos del complemento. Los reguladores de superficie controlan a la C3 convertasa a través de la inactivación del C3b depositado en la superficie celular y la membrana basal.
La desregulación de la vía alternativa puede producirse debido a las mutaciones de los factores del complemento o por autoanticuerpos contra las proteínas reguladoras del complemento o C3 convertasa. Por ejemplo, las mutaciones en las proteínas que regulan el ensamblaje y la actividad de la C3 convertasa y la degradación de C3b, tales como los factores H, I y B y la proeteína 5 relacionada con el factor H, da como resultado la desregulación de las vías alternativas. Las mutaciones heterocigotas del C3 causan la desregulación en la fase líquida de la vía alternativa, ya que el C3 mutante es resistente a la escisión por la C3 convertasa. Por otra parte, la generación a través del mecanismo “a ralentí” de una C3 convertasa anormal, que contiene la mutación C3, la hace resistente a la inactivación por el factor H. A continuación, la C3 convertasa anormal se unirá al C3 producido por el alelo C3 normal, dando como resultado mayores niveles de productos derivados de C3. Del mismo modo, los anticuerpos del de las proteínas reguladoras del complemento (tales como los factores H y B) y la C3-convertasa en sí misma pueden provocar la hiperactividad de las vías alternativas. Los anticuerpos contra la C3 convertasa (llamada factor nefrítico C3) estabilizan la convertasa y prolongan su vida media previniendo su inactivación y degradación y produciendo la hiperactivación de la vía alternativa.
Ciertos polimorfismos genéticos en los factores H y B, la proteína cofactor de membrana y C3 también se asocian con la GNMP. Los polimorfismos en el gen que codifica el factor H, en particular las variantes del alelo Tyr402His, son los polimorfismos estudiados con más frecuencia. En comparación con Tyr402, His402 está excesivamente representado en los pacientes con GNMP y las anormalidades de la vía alternativa; los estudios funcionales muestran que His402 afecta la regulación de la C3 convertasa mediada por el factor H en la superficie celular.
Cualquiera sea el mecanismo, la desregulación de la vía alternativa da como resultado la formación de productos del complemento activados, incluyendo C3b y los factores del complemento terminal, que son liberados de manera indiscriminada a la superficie endotelial, incluyendo los glomérulos. La deposición de estos productos del complemento y residuos en el mesangio y la región subendotelial provoca la inflamación glomerular y conduce a la GNMP. Las inmunoglobulinas no están directamente implicadas, por lo que en los estudios de inmunofluorescencia, la GNMP mediada por el complemento es típicamente inmunoglobulina-negativa, pero complemento-positiva.
A pesar de los múltiples factores de riesgo genéticos, la GNMP secundaria a las anomalías del complemento a menudo se desarrolla relativamente tarde en la vida, lo que sugiere que son obligatorios los insultos adicionales o los factores ambientales. Por otra parte, la GNMP no se desarrolla en todos los miembros genéticamente similares de familias de alto riesgo, indicando de nuevo que se necesitan otros factores adicionales que inducen la enfermedad. Los autores especulan que cuando la GNMP no ocurre a pesar de la presencia de una mutación o de un alelo variante que confiere una predisposición a la enfermedad, puede haber mecanismos de control redundantes. Sin embargo, cuando se produce un insulto adicional como una infección o la activación del complemento, se puede saturar el mecanismo de regulación compensatorio, provocando la deposición glomerular de factores del complemento. Esta situación puede explicar los episodios recurrentes de hematuria macroscópica asociada a las infecciones (hematuria sinfaringítica), observada en muchos pacientes con GNMP. Del mismo modo, un insulto adicional como la producción de proteínas monoclonales que actúan como autoanticuerpos contra las proteínas reguladoras del complemento en pacientes con GMSI podría provocar la desregulación de la vía alternativa y el desarrollo de MPGN.
Características anatomopatológicas
La deposición de inmunoglobulinas, complemento, o ambos en la región del mesangio y subendotelial de la pared capilar provoca una lesión aguda que a menudo es seguida por una fase inflamatoria (celular o proliferativa), con la afluencia de células inflamatorias. Posteriormente, se produce una fase de reparación, durante la cual la producción de matriz mesangial nueva provoca la expansión del mesangio, junto con la generación de membrana basal glomerular nueva, que se ve como una membrana basal duplicada (denominada “vías del tranvía” o contornos dobles).
Los hallazgos con inmunofluorescencia se utilizan para distinguir la GNMP mediada por complejos inmunes de la GNMP mediada por el complemento y puede apuntar a menudo a una causa específica. Por ejemplo, la GNMP asociada a la gammapatía monoclonal muestra inmunoglobulina monotípica con restricción de cadenas livianas kappa o lambda. La GNMP asociada con la infección por hepatitis C generalmente muestra IgM, IgG, C3, y cadenas livianas kappa y lambda. El patrón de GNMP asociada a enfermedades autoinmunes suele incluir inmunoglobulinas y múltiples proteínas del complemento—IgG, IgM, IgA, C1q, C3 y cadenas livianas kappa y lambda. La GNMP asociada a la disfunción de vía alternativa se caracteriza por la inmunotinción brillante de C3 en el mesangio y a lo largo de las paredes de los capilares. La ausencia de tinción de la inmunoglobulina marcada en el microscopio de inmunofluorescencia distingue a la GNMP por disfunción de la vía alternativa de la GNMP mediada por complejos inmunes.
La microscopía electrónica muestra depósitos mesangiales y subendoteliales y, en algunos casos, depósitos intramembranosas y subepiteliales y durante la fase reparadora, formas nuevas de membrana basal, depósitos atrapados en la pared capilar junto con elementos celulares derivados de las células inflamatorias, mesangiales y endoteliales en el material de la nueva membrana basal. El resultado es un engrosamiento de las paredes de los capilares y la formación de dobles contornos a lo largo de las paredes de los capilares. Con la excepción de la enfermedad de depósito denso, la microscopía electrónica no puede distinguir entre la GNMP mediada por complejos inmunes y la GNMP mediada por el complemento.
Según los hallazgos de la microscopía electrónica, la GNMP debida a la desregulación de la vía alternativa se puede subdividir en la enfermedad con depósitos de alta densidad y la glomerulonefritis C3 (GNC3). La enfermedad con depósitos de alta densidad se caracteriza por depósitos densos ondulados osmiófilos, en forma de salchicha, que reemplazan a la membrana basal glomerular y también se producen en el mesangio, mientras que GNC3 tiene depósitos mesangiales, subendoteliales , subepiteliales y a veces intramembranosos. Sobre la base de las características morfológicas de la GNC3 en la microscopía electrónica, es más probable que la GNC3 se denomine GNMP I o III de acuerdo con la clasificación mayor de GNMP. Los datos de la microdisección láser y el análisis de la espectrometría de masas de los glomérulos obtenidos de pacientes con GNC3 son consistentes con la activación no restringida de la vía alternativa, y el perfil proteómico en estos pacientes es similar al de los pacientes con enfermedad con depósitos de alta densidad. La hipótesis de que la enfermedad con depósitos de alta densidad y la GNC3 son parte de un continuum está más apoyada por los casos que muestran características intermedias entre la enfermedad con depósitos de alta densidad y la GNC3, mostrando en algunas asas capilares la presencia de depósitos intramembranosos en forma de salchicha de la enfermedad de depósito denso y otros bucles que muestran los depósitos subendoteliales y subepiteliales de GNC3 en la microscopía electrónica.
Desregulación de la vía alternativa y subtipo de enfermedad
En algunos pacientes, la desregulación de la vía alternativa resulta en la enfermedad de depósitos densos pero en otros, el resultado es la GNC, muy probablemente debido a las diferencias en el grado o el sitio de la desregulación (o ambos). Por otra parte, ciertas variaciones alélicas de las proteínas reguladoras del complemento pueden estar asociadas a la enfermedad de depósitos densos, y otras pueden estar asociadas a la GNC3.
Otros patrones de lesiones glomerulares mediadas por complejos inmunes y el complemento
La deposición de inmunoglobulina, complemento, o ambos, puede ocasionar otros patrones de lesión glomerular además de la GNMP. Por ejemplo, tanto en la GNC3 como en la enfermedad de depósito denso pueden presentarse como una glomerulonefritis proliferativa mesangial, glomerulonefritis proliferativa difusa, glomerulonefritis y glomerulopatía esclerosante.
El término general " glomerulopatía C3" describe distintos patrones de lesión que probablemente dependen de múltiples factores, entre ellos la gravedad de las lesiones y la fase del proceso de la enfermedad (aguda o crónica) en el momento que se lleva a cabo la biopsia. El tratamiento previo también puede afectar los resultados de la biopsia.
GNMP sin complejos inmunes o complemento
Un patrón de lesiones consistente con la GNMP también es el de las microangiopatías trombóticas derivadas de una lesión a las células endoteliales. En la fase aguda se observa mesangiólisis, inflamación endotelial y trombos de fibrina en los capilares glomerulares. Dado que el proceso pasa a una fase reparadora y crónica, tiene lugar la expansión mesangial y la remodelación de las paredes capilares glomerulares, incluyendo la formación de doble contorno. Así, la curación de la fase de púrpura trombocitopénica trombótica o síndrome urémico hemolítico o síndrome urémico hemolítico atípico se asoció a las alteraciones del complemento, el síndrome de anticuerpos antifosfolípidos, las microangiopatías trombóticas inducidas por fármacos, la nefropatía asociada al trasplante de médula ósea, la nefritis por radiación, la hipertensión maligna y los trastornos del tejido conectivo, los cuales pueden presentarse con una lesión con patrón de GNMP en la biopsia. En las microangiopatías trombóticas, los hallazgos de la microscopia electrónica se caracterizan por la ausencia inmunofluorescencia de las inmunoglobulinas y el complemento la, como así de depósitos densos en el mesangio o a lo largo de las paredes de los capilares.
Evaluación
En los pacientes con GNMP comúnmente se observa la persistencia de la disminución de los niveles séricos de complemento C3, C4, o ambas cosas. La presencia de niveles bajos de complementos C3 y C4 es más común en la GNMP mediada por complejos inmunes, mientras que el C3 bajo con niveles normales de C4 es más común en la GNMP por disfunción de la vía alternativa, particularmente en la fase aguda. Un nivel normal de C3 no descarta la disfunción de la vía alternativa.
Cuando una muestra de biopsia renal de un paciente con GNMP muestra inmunoglobulinas está indicado investigar la presencia de infecciones, enfermedades autoinmunes o gammapatías monoclonales. Las pruebas pertinentes para la detección de infecciones incluyen los hemocultivos y la reacción en cadena de la polimerasa, además del análisis serológicos para bacterianas, virus y hongos. Pueden hallarse crioglobulinas. Las pruebas para la detección de las gammapatías monoclonales incluyen la electroforesis del suero y la orina, los estudios de inmunofijación y el análisis de las cadenas livianas libres; los resultados positivos requieren estudios de la médula ósea para arribar a un diagnóstico más preciso. Las pruebas positivas de detección de una enfermedad autoinmune deben ser seguidas por las pruebas específicas para la enfermedad autoinmune.
Si la muestra de biopsia de un paciente con GNMP muestra inmunotinción C3 brillante (con una tinción de inmunoglobulina mínima o nula), deben iniciarse estudios para detectar anomalías de la vía alternativa, independientemente de si se hallan signos de enfermedad de depósitos densos o de GNC3 en la microscopía electrónica. La evaluación inicial de la vía alternativa debe incluir la medición de los niveles séricos del complemento y del complejo de ataque de membrana, un análisis funcional de la vía alternativa y ensayos del complemento hemolítico, seguidos del análisis genéticos de las mutaciones y variantes alélicas de los factores del complemento y los análisis para detectar la presencia de autoanticuerpos contra las proteínas reguladoras del complemento, incluyendo las pruebas para la detección del factor nefrítico C3.
Incluso después de una extensa evaluación, algunos casos de GNMP mediada por complejos inmunes o mediada por complemento que pueden seguir siendo enigmáticos. En algunos pacientes, la GNMP mediada por complejos inmunes puede comenzar con la deposición de inmunoglobulinas, pero la enfermedad puede estar acelerada por las anomalías de la vía alternativa. Es probable que con el tiempo se desarrollen métodos
nuevos que puedan separar más las causas específicas de la GNMP de los casos idiopáticos.
Tratamiento
Los primeros informes sobre el tratamiento de la GNMP "idiopática" deben ser interpretados con precaución. En muchos casos, se utilizaron los controles históricos, la significación estadística fue marginal o el poder para detectar las diferencias sustanciales fue pequeño. La mayoría de los estudios fueron anteriores al uso de los inhibidores de la enzima convertidora de angiotensina y bloqueantes de los receptores de la angiotensina II, y a al conocimiento de los procesos patogénicos proteicos que conducen a la GNMP. Por lo tanto, la mayoría de los estudios sobre la GNMP reunieron varios tipos de GNMP en una proporción desconocida.
Algunos estudios no controlados y un estudio controlado aleatorizado han mostrado el beneficio a largo plazo del tratamiento con glucocorticoides en días alternos de los niños con GNMP idiopática. Sin embargo, estos estudios incluyeron una mezcla de pacientes con GNMP I, GNMP III y enfermedad de depósitos densos, limitando las conclusiones a las que se podía llegar. No ha habido una evaluación sistemática del tratamiento con glucocorticoides para la GNMP idiopática en adultos. Los estudios retrospectivos no mostraron ningún beneficio claro del tratamiento con glucocorticoides, pero el tratamiento no fue tan prolongado en los adultos como lo fue en los niños.
Los primeros informes de los efectos beneficiosos de los anticoagulantes (heparina y warfarina), frecuentemente combinados con glucocorticoides y agentes citotóxicos, no han sido confirmados en un estudio prospectivo. Del mismo modo, uno de los primeros estudios aleatorizado y controlado mostró que la combinación de aspirina y dipiridamol produjo un descenso más lento de la tasa de filtración glomerular en los adultos con GNMP idiopática, pero sin ningún beneficio a largo plazo, lo que sugiere que para lograr un beneficio sostenido es necesaria la antiagregación prolongada. Los pocos datos no controlados existentes sugieren que en algunos pacientes con GNMP, los inhibidores de la calcineurina pueden reducir la proteinuria. En los pacientes con un curso rápidamente progresivo y medias lunas en la biopsia renal, unos pocos estudios pequeños no controlados han sugerido un beneficio con “pulsos” con dosis elevadas de glucocorticoides, ya sea como monoterapia o en combinación con azatioprina, ciclofosfamida o micofenolato mofetil.
La falta de estudios aleatorizados y controlados, y el conocimiento actual de la existencia de múltiples procesos patogénicos que conducen a la GNMP hacen imposible dar recomendaciones precisas sobre el tratamiento en esta población de pacientes. Las consideraciones pragmáticas sugieren que los pacientes con GNMP por infecciones crónicas deben someterse a un tratamiento de la infección, y aquellos con GNMP debida a una enfermedad autoinmune deben someterse a un tratamiento de la enfermedad autoinmune. Del mismo modo, los pacientes con GNMP debido a una gammapatía monoclonal deben recibir tratamiento dirigido a lograr la remisión de la discrasia hematológica. Un estudio reciente con pacientes con GNMP asociada a depósitos de inmunoglobulina monoclonal y sin cáncer hematológico manifiesto mostró que los pacientes tenían una buena respuesta al rituximab. Los pacientes con niveles normales de la función renal, sin sedimento urinario activo y sin proteinuria en rango nefrótico pueden ser tratados de forma conservadora con bloqueantes de la angiotensina II, para controlar la presión arterial y reducir la proteinuria, ya que en este contexto el resultado a largo plazo es relativamente benigno. Es necesario hacer el seguimiento para detectar el deterioro prematuro de la función renal. Una mejor comprensión de las causas y patogénesis de la GNMP mediada por el complemento sentaría las bases para el posible uso de nuevos fármacos, incluyendo las drogas anticomplemento. Sin embargo, las recomendaciones actuales están basadas en la teoría y no en estudios. Por ejemplo, los pacientes con GNMP por autoanticuerpos contra las proteínas reguladoras del complemento pueden beneficiarse con el tratamiento inmunosupresor (por ej., glucocorticoides y rituximab), mientras que aquellos con GNMP debido a una mutación genética en las proteínas reguladoras del complemento pueden beneficiarse del tratamiento con fármacos que inhiben la formación del complejo de ataque a la membrana (por ej., eculizumab). El eculizumab, un anticuerpo monoclonal anti-C5 que inhibe la activación de C5, se ha utilizado con éxito en los pacientes con síndrome urémico hemolítico atípico secundario a anormalidades en la vía alternativa del complemento. El papel de tales agentes anticomplemento en la GNMP no está delineado pero ofrece interesantes posibilidades para el futuro. También es concebible que respondan al eculizumab los pacientes con niveles séricos elevados del complejo de ataque a la membrana que aquellos con niveles normales. Los pacientes con GNMP debido a una deficiencia del factor H podrían beneficiarse con la infusión de plasma o de factor H. Es poco probable que los pacientes que se presentan con insuficiencia renal avanzada severa y fibrosis tubulointersticial en la biopsia renal se beneficien con el tratamiento inmunosupresor.
Recurrencia después del trasplante renal
La GNMP se repite a menudo en los pacientes con trasplante de riñón. Las tasas de recurrencia oscilan entre el 27 y el 65%, dependiendo del estudio. Un ensayo reciente que excluyó del análisis a los pacientes con enfermedad de depósitos densos mostró una tasa de recurrencia del 41%; de los pacientes con una recurrencia, el 36% tenía una gammapatía monoclonal. El estudio demostró que la GNMP recurrente debido a la deposición de inmunoglobulina monoclonal se asoció con la recidiva precoz y una evolución más agresiva. Los niveles de complemento se mostraron como un marcador precoz de GNMP recurrente Existen pocos datos sobre la recurrencia de la GNC3. En los pacientes con enfermedad de depósitos densos, es casi universal la recurrencia de la enfermedad, con una tasa de fracaso del injerto a los 5 años del 50%.
Conclusiones
Dos son los factores principales que pueden conducir a la GNMP: la deposición fisiopatológica de inmunoglobulina y la deposición del complemento en el mesangio glomerular y las paredes capilares. La presencia de GNMP mediada por complejos inmunes requiere la evaluación de las infecciones, las enfermedades autoinmunes y la gammapatía monoclonal. La GNMP mediada por el complemento se subdivide en enfermedad de depósitos densos y GNC3, dependiendo de los hallazgos en el microscopio electrónico; la presencia de GNMP mediada por el complemento requiere la evaluación de la vía alternativa. La evaluación de la GNMP de acuerdo con el proceso fisiopatológico subyacente puede facilitar el tratamiento adecuado.
♦ Traducción y resumen. Dra. Marta Papponetti.
♦ Para acceder a las referencias bibliográficas en formato Pdf, haga clic aquí