El linfoma intravascular (IVL) es un subtipo de linfoma No-Hodgkin extranodal, con una incidencia estimada de menos de 1 caso por millón de personas. Se caracteriza por una extensa proliferación de células de linfoma en vasos sanguíneos de pequeño y mediano tamaño. La mayoría de los IVL son tumores de células B. Se pueden presentar en cualquier órgano o sistema incluyendo la piel. La enfermedad es generalmente diseminada al momento del diagnóstico. Se piensa que el porcentaje de mortalidad es mayor al 80% y el 50% de los pacientes se diagnostican postmortem. El IVL puede presentarse con un rash cutáneo que precede o acompaña otros signos y síntomas de la enfermedad. La apariencia clínica de las lesiones cutáneas de IVL varía ampliamente.
Se reportan dos pacientes con enfermedad sistémica inexplicable y erupción cutánea, con diagnóstico de IVL y se detalla el rango de características cutáneas reportadas.
Reporte de casos.
Paciente 1.
Mujer de 59 años, con dolor agudo abdominal con sospecha de obstrucción de intestino delgado. Los antecedentes incluían artritis reumatoidea por lo que recibía sulfazalacina 1.5 g dos veces por día. Se resecó un segmento isquémico del íleon distal; al exámen histológico se observó trombosis e infarto, pero sin causa específica identificable. Tres semanas después, la paciente desarrolló un rash eritematoso indurado, simétrico y doloroso asociado con telangiectasias prominentes sobre los flancos, muslos y abdomen inferior (fig 1a). No presentaba organomegalias palpables. El laboratorio reveló anemia normocítica inexplicable Hb 8.3 g/dl. La tomografía computada de abdomen sin particularidades. En el exámen histológico de la biopsia de piel (fig 1b), se observaban linfocitos grandes atípicos en capilares y vasos sanguíneos del subcutis. Con la inmunohistoquímica, éstas células eran positivas al antígeno común leucocitario (LCA), CD5, CD20 y CD79a. Basado en éstos hallazgos se realizó el diagnóstico de IVL de células B grandes. Por lo que se revisó la pieza de íleon distal, y se observaron infiltrados de linfocitos atípicos, en paredes de los vasos en submucosa y tejido subcutáneo. No presentaba compromiso de médula ósea y la tomografía de emisión de positrones corporal total no identificó lesiones. Se inició tratamiento con R-CHOP (rituximab, vincristina, doxorrubicina, ciclofosfamida, y prednisolona), y luego de 3 ciclos de tratamiento, el compromiso cutáneo había resuelto clínicamente, y el paciente entró en remisión durante 13 meses. Sin embargo, la enfermedad recayó con linfadenopatía generalizada y metástasis óseas. A pesar de la quimioterapia y radioterapia paliativa, la paciente falleció 9 meses después.
.jpg)
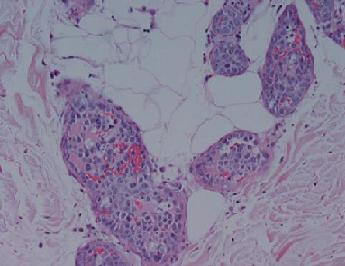
Figura 1 Paciente 1. (a) Placas induradas eritematosas telangiectàsicas en abdomen y piernas (b) infiltrado de células grandes atípicas, mitóticamente activas, con espacios vasculares distendidos de la dermis y tejido subcutáneo (H&E).
Paciente 2.
Hombre de 76 años, que presentaba un rash en piernas de 3 meses de evolución. Era doloroso, livedoide, simétrico y eritematoso (fig 2a). También presentaba malestar general, pirexia, pérdida de peso, anorexia y sudoración nocturna. Presentaba esplenomegalia detectable clínicamente pero no presentaba hígado palpable ni adenomegalias. Fue tratado empíricamente con antibióticos de amplio espectro, aunque no se identificaron focos de infección. Presentó deterioro y confusión.
Los marcadores de inflamación estaban elevados, eritrosedimentación de 66 mm/h, PCR 150mg/L, y LDH sérica de 480 UI/L. En la tomografía de cerebro se observó una masa intraventricular en el ventrículo lateral derecho, y el paciente fue transferido a la unidad neuroquirúrgica. En la biopsia de piel del rash de pierna izquierda (fig 2b), se observaron linfocitos atípicos en los vasos sanguíneos. Con la inmunotinción éstas células fueron positivas para LCA, CD20, y CD79a, confirmando el diagnóstico de IVL de células B. La tomografía para estadificación no mostró masas intraabdominales ni torácicas de nódulos linfáticos. No se realizó biopsia de la lesión intraventricular. El paciente fue tratado con R-CHOP. La mejoría cutánea se observó luego de dos ciclos de quimioterapia y se logró remisión del linfoma luego de seis ciclos, con un hemograma completo normal y linfoadenopatías no palpables. Una tomografía y resonancia magnética de cerebro no mostraron cambios en la masa intraventricular, que se consideró como una patología no relacionada, posiblemente un subependimoma. El paciente permaneció bien y seguía en remisión 45 meses posteriores al diagnóstico de IVL.
El IVL o linfoma angiotrópico (antes llamado angioendoteliomatosis maligna), fue primeramente descripto por Pfleger y Tappeiner. Es un linfoma extranodal raro, que se diagnostica patológicamente por la acumulación y proliferación clonal de linfocitos B grandes neoplásicos en el lúmen de vasos sanguíneos pequeños y medianos, ocasionando oclusión vascular. La enfermedad es reconocida como un subtipo de linfoma difuso de células B grandes según la clasificación de la Organización Mundial de la Salud (WHO), aunque existen formas raras con fenotipo de células T. La etiología de ésta condición es desconocida, aunque se han sugerido asociaciones con el virus de Epstein_barr y virus de linfocitos de células T humano, en casos individuales. La mayoría de los pacientes mueren 1 año luego del diagnóstico por compromiso de órganos secundarios.
En la serie de casos más grande publicada, el compromiso de la piel se observó como característica mayor en 15 de 38 casos (39%). De ellos 4 casos tenían lesiones solitarias y 11 casos lesiones múltiples. La piel fue el único sitio comprometido en 10 pacientes, pero como en el segundo caso presentado, se asociaban a síntomas neurológicos y tipo B en 5 pacientes.
Se han reportado varias lesiones cutáneas en IVL. Estas incluyen erupciones morbiliformes, nódulos, placas, telangiectasias, parches hiperpigmentados, púrpura palpable y úlceras. También se ha reportado la colonización de hemangiomas cutáneos por células neoplásicas, como único signo de IVL. Las lesiones predominan en superficies proximales de los miembros, abdomen inferior y regiones submamarias. No se encontró relación entre el tipo de lesión cutánea y otro compromiso orgánico. Las lesiones de IVL en la piel pueden simular varias enfermedades como tromboflebitis migratoria, eritema nodoso, erisipela, vasculitis o vasculopatía oclusiva secundaria a trombofilia. Luego del exámen clínico, el IVL puede no ser parte de diagnósticos diferenciales dermatológicos. Esto subraya la importancia de realizar una biopsia de piel para asegurar el diagnóstico.
En la mayoría de los casos el IVL es diseminado al momento del diagnóstico y requiere tratamiento sistémico. El porcentaje de sobrevida a los 3 años luego del tratamiento R-CHOP se estima que es del 32+-11%.
En conclusión, se reportaron dos pacientes con IVL de células B, que se presentaron a cirujanos vasculares o generales inicialmente, pero fueron diagnosticados luego de examinar las lesiones cutáneas y realizar biopsia de piel. El conocer las manifestaciones cutáneas de IVL puede permitir un diagnóstico más temprano de éste linfoma raro y altamente agresivo. Esto es importante, ya que cuando el diagnóstico se realiza y se limita a la piel confiere un mejor pronóstico que si la enfermedad está diseminada.
.jpg)
.jpg)
Figura 2 Paciente 2. (a) Rash macular eritematoso, livedoide en ambas piernas; (b) infiltrado de linfocitos atípicos en espacios vasculares de la dermis (H&E).
¿Qué aporta éste artículo a la práctica dermatológica?
El linfoma intravascular (IVL) es un subtipo de linfoma no-Hodgkin extranodal, con una incidencia estimada de menos de 1 caso por millón de personas. Se caracteriza por proliferación extensa de células de linfoma en vasos sanguíneos de pequeño a mediano tamaño. La mayoría de IVL son tumores de células B. El linfoma intravascular puede presentarse primariamente en cualquier órgano, incluyendo la piel. La enfermedad es generalmente diseminada al momento del diagnóstico. El porcentaje de mortalidad se piensa que es mayor al 80% y el 50% de los pacientes se diagnostican postmortem. Existe una amplia variabilidad en la apariencia clínica de las lesiones cutáneas, que pueden simular enfermedad inflamatoria cutánea. Por lo que es importante el diagnóstico temprano cuando están presentes los signos cutáneos. Se reporta el caso de dos pacientes con enfermedad sistémica inexplicada y erupción cutánea, diagnosticándose IVL.
♦ Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello.