Mieloma múltiple

| Etiología/fisiopatología |
El mieloma múltiple (MM) se caracteriza por la acumulación de células plasmáticas clonales malignas en la médula ósea.
La causa es desconocida; en algunos casos, puede estar provocado a la radiación (aunque no hay asociación con la radiación terapéutica), la exposición a toxinas industriales o agrícolas, y también se ha propuesto una asociación con los virus pero faltan pruebas.
Se han identificado anomalías cromosómicas, más comúnmente involucrando al interruptor de la región de cadena pesada de inmunoglobulinas (Ig) (en el brazo largo del cromosoma 14), aunque esto. por sí solo, no parece ser suficiente para dar lugar al MM).
Las células tumorales en la médula ósea están soportadas por una población de células estromales no malignas que producen citocinas (p. ej., interleucina-6), cuya acción potencia el crecimiento celular del mieloma y previene la apoptosis.
| Epidemiología |
El MM representa casi el 10% de las neoplasias hematológicas. La incidencia anual en el Reino Unido es de 5/100.000. Las personas de raza negra son afectadas 2 veces más comúnmente que las personas de raza blanca, y los hombres más que las mujeres
La edad media al momento del diagnóstico es de 65 años. La presentación en menores de 40 años ocurre en menos del 3% de los pacientes.
| Presentación clínica |
> Común
• Dolor óseo y fracturas patológicas.
• Anemia (insuficiencia de la médula ósea)
• Infecciones recurrentes (por inmunoparesia)
• Hipercalcemia
• Insuficiencia renal (etiologías múltiples: hipercalcemia, deposición de cadenas livianas, medicamentos no esteroideos, anemia, infecciones)
• Sangrado anormal (debido a disfunción plaquetaria)
> Rara
• Síndrome de hiperviscosidad (isquemia, insuficiencia cardíaca y problemas neurológicos)
• Enfermedad amiloide (por ej., síndrome del túnel carpiano)
| Investigaciones |
Ante la sospecha de MM, pueden ser útiles las siguientes pruebas:
• Hemograma completo: puede revelar anemia normocrómica, normocítica.
• Velocidad de sedimentación elevada (la carga positiva de proteína neutraliza la carga negativa del ácido siálico en la membrana eritrocítica, reduciendo su tendencia natural a rechazarse entre sí y haciendo que las células se depositen más rápido en una columna).
• Determinación de la hipercalcemia (generalmente con una fosfatasa alcalina normal) y alteración renal.
• Electroforesis sérica: puede demostrar una proteína monoclonal, que en la mayoría de los casos es IgG o IgA, pero puede ser cualquier clase de Ig. Los niveles reducidos de Ig normales son confirmatorios.
• En el suero puede hallarse el exceso de cadenas livianas libres, ya sea kappa o lambda (a veces el clon de células plasmáticas es solo de cadenas livianas).
• El diagnóstico de MM depende de la demostración del aumento de células plasmáticas (>10%) en la médula ósea.
• Las radiografías óseas pueden mostrar lesiones líticas.
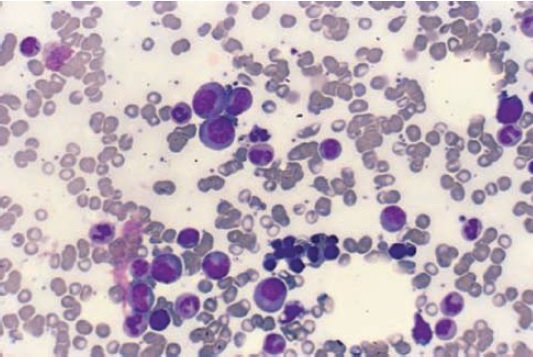
Células de mieloma. Esta médula ósea muestra diferentes tipos de células. Las células más grandes con núcleos excéntricos y citoplasma basófilo son células de mieloma. Tenga en cuenta la transparencia perinuclear que representa el aparato de Golgi.
| Estadificación (Sistema Internacional de Estadificación) |
La estadificación se relaciona con los niveles de albúmina y la proteína β2-microglobulina.
| Signos del mieloma | ||
| Estadio 1 | Estadio 2 | Estadio 3 |
| ß2-Ig < 3,5 mg/dl | Ni 1 ni 3 | ß2-Ig >5,5 mg/dl |
| Albúmina >35 g/l | ||
| Diagnóstico diferencial de una paraproteína sérica |
Maligno
• Macroglobulinemia de Waldenström
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.