Actualización de la genética y expresión clínica de poroqueratosis

Se presenta el caso de una mujer coreana de 73 años con antecedentes de artritis reumatoidea, hipertensión, diabetes tipo 2, síndrome de intestino irritable, y episodios previos de pancreatitis aguda que presentó lesiones cutáneas de larga evolución en extremidades, monte de venus en pubis y glúteos. En las extremidades distales (fig 1a, brazo y b piernas), presentaba parches y placas atróficas pequeñas numerosas bilaterales de 2 a 5 mm de diámetro rodeadas de crestas de queratina pigmentadas que son características de poroqueratosis actínica superficial diseminada. El aspecto lateral de los muslos (fig 1 c) presentaba numerosas pápulas pequeñas, marrones, verrugosas de 1 a 2 mm con una distribución linear segmentaria bilateral. Ambos tipos de lesiones de las extremidades han estado presentes por al menos 10 años y tienen inicio separado. En los glúteos y monte de venus (fig 1 d glúteos), presentaba placas grandes, bilaterales, bien delimitadas hiperpigmentadas en la hendidura del área glútea, con pápulas que se extendían a la parte interna de los muslos y monte de venus. Estas aparecieron al menos hace 30 años. Los 3 tipos de lesiones eran bastante diferentes clínicamente.
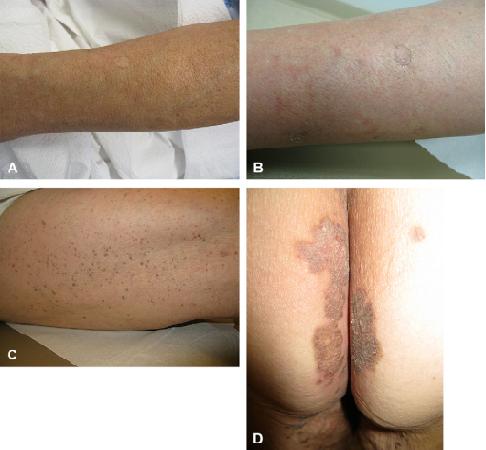
Fig 1. Imágenes clínicas de pacientes con DSAP coexistente. Brazo distal (A) y pierna distal (B) en extremidades, poroqueratosis linear en la parte lateral de muslos (C) y poroqueratosis verrucosa (D) en glúteos.
El hijo de la paciente desarrolló pequeñas pápulas verrucosas en el lateral de los muslos, pero no tenía placas grandes verrucosas en sus glúteos o de lesiones de poroqueratosis actínica superficial diseminada (DSAP) en las extremidades distales.
Hallazgos histopatológicos:
Las biopsias se tomaron del área de extremidades distales, parte lateral del muslo, y monte de venus con morfología similar a la de los glúteos. De la extremidad distal (fig 2a) una epidermis delgada atrófica contenía lamelas corneoides infrecuentes, que se ve como una columna de paraqueratina encima de una invaginación focal de epidermis con pérdida de la granulosa y queratinocitos apoptóticos.
En la parte lateral del muslo (fig 2b), la epidermis contenía múltiples lamelas corneoides por campo. La dermis contenía incremento de vasos pequeños y parches de infiltrados de células mononucleares. Los hallazgos de la biopsia de monte de venus y glúteos eran similares (fig 2c).
La epidermis hiperplásica contenía numerosas lamelas corneoides amplias, en algunas áreas casi contiguas. Debajo de la lamela corneoide había ausencia de granulosa y queratinocitos apoptóticos. La biopsia de los 3 sectores, extremidad distal, parte lateral de muslo y monte de venus, tenían incremento de pequeños vasos y de infiltrado de células mononucleares en dermis papilar, más pronunciados en muslo y monte de venus. Las lesiones de muslo y monte de venus tenían caída de pigmento en dermis superior, de acuerdo con la hiperpigmentación clínica.
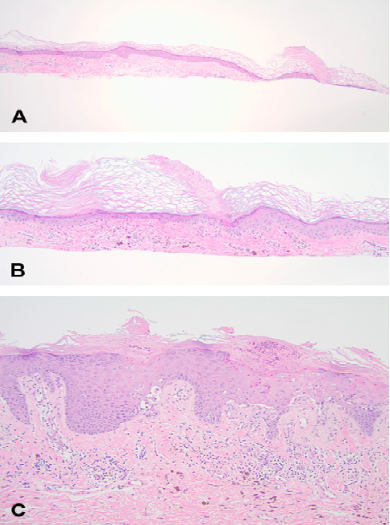
Fig 2. Imágenes representativas de características histopatológicas para fig 1, B-D. A, extremidades distales, biopsia que muestra DSAP clásica. Una lamela corneoide está flanqueada por epidermis atrófica.B, Muslo: biopsia de poroqueratosis linear muestra varias lamelas corneoides por campo, infiltrado mononuclear liquenoide en dermis papilar, y caída de pigmento. C, biopsia de poroqueratosis verrucosa de monte de Venus que muestra múltiples lamelas corneoides con epidermis hiperplásica acantótica- al menos hay dos en ésta imagen- como así también pequeñas lamelas corneoides clásicas. Inflamación liquenoide y caída de pigmento en dermis papilar que se correlacionan con la hiperpigmentación clínica. Los hallazgos observados en los glúteos eran similares.
(A-C, Hematoxilina-eosina)
Características clínicas:
Los factores de riesgo para DSAP, la forma más común y estudiada de poroqueratosis incluye factores genéticos, exposición ultravioleta, e inmunosupresión. Existe un incremento del 7.5% al 10% en poroqueratosis-principalmente poroqueratosis de Mibelli y poroqueratosis superficial diseminada, una variante similar a DSAP que ocurre en piel no expuesta al sol- en individuos inmunosuprimidos por la reducida inmunidad. Estos factores de riesgo incluyen transplantes de órganos, linfomas, infección por HIV, y enfermedades inflamatorias o autoinmunes tratadas con inmunosupresores, con incidencia más alta en trasplantados de órganos sólidos, principalmente de riñones. En la paciente estudiada la enfermedad intestinal y episodios de pancreatitis pueden haber predispuesto al desarrollo de poroqueratosis, aunque no estaba recibiendo inmunosupresores.
Sorprendentemente, no hay incremento de cáncer cutáneo que se desarrolle de lesiones de poroqueratosis en individuos que fueron sometidos a transplante de órganos, que tienen riesgo más alto de desarrollar cáncer cutáneo de otro modo.
En una revisión de 281 individuos con poroqueratosis desde 1964 a 1994, la incidencia más alta de carcinomas cutáneos fue en pacientes con poroqueratosis linear (19%). La incidencia de carcinoma en las otras formas era la siguiente: poroqueratosis de Mibelli (7.6%), DSAP (3.4%), poroqueratosis palmar y plantar (9.5%) y poroqueratosis punctata (0%). Ninguno de los individuos con carcinoma originado de poroqueratosis estaba iatrogénicamente inmunosuprimidos. Un individuo presentaba síndrome de Bloom y uno síndrome de Werner. Los carcinomas se presentaban en individuos de mediana edad con lesiones grandes y enfermedad de larga duración.
Las malignidades eran principalmente carcinomas de células escamosas o enfermedad de Bowen (21 de 23 carcinomas) con un pequeño número de carcinoma de células basales.
Coexistencia de formas variadas de poroqueratosis:
Existen reportes de poroqueratosis verrucosa en glúteos simulando psoriasis clínicamente como así también la coexistencia de múltiples tipos de poroqueratosis en el mismo individuo. Cuando un individuo tiene poroqueratosis linear aislada, generalmente hay miembros de la familia con DSAP. La paciente en estudio tenía DSAP en las extremidades distales, poroqueratosis linear en la parte lateral de muslos y poroqueratosis verrucosa en glúteos y monte de venus en pubis. Aunque la apariencia clínica de las lesiones en muslos no eran pápulas lineares, se presentaban pápulas agrupadas en una distribución segmentaria. La placa en los glúteos y monte de venus no era verrucosa clínicamente ni histológicamente, pero la localización y apariencia se asemeja a otras descripciones clínicas publicadas de poroqueratosis verrucosa.
Histopatología:
La característica diagnóstica de todas las formas de poroqueratosis es la lamela corneoide. Se ha descripto el incremento de múltiples lamelas corneoides grandes en la poroqueratosis linear. Se sugiere disregulación de proliferación de múltiples clones de queratinocitos contiguos con expresión más dramática que en DSAP, en la que se puede identificar sólo una lamela corneoide en la epidermis luego de varios cortes.
Genética:
La genética de la poroqueratosis nos se ha resuelto completamente, pero se han propuesto varias explicaciones para las manifestaciones de la poroqueratosis. La DSAP se hereda de manera autosómica dominante con disminución de la penetrancia del 22%. Un alelo defectuoso en el genoma de un individuo heterocigocico es suficiente para expresar la enfermedad. La expresión cutánea es difusa, porque todas las células del cuerpo tienen la mutación. La expresión retardada de DSAP en el adulto es probablemente por eventos de mutación gatillados por la exposición a largo plazo. Las formas que se presentan en la infancia y niñez (poroqueratosis de Mibelli y poroqueratosis linear) probablemente tengan genéticas diferentes. Durante el desarrollo embriológico, la mutación somática que causa pérdida de la heterocigocidad puede ocurrir en células que se diferencian al azar incluyendo queratinocitos, produciendo un individuo que es un mosaico por el defecto en dos alelos y tiene parches en las líneas de Blascho de células homocigotos defectuosas.
Happle ha propuesto que la recombinación somática puede explicar la coexistencia de múltiples formas de poroqueratosis como la poroqueratosis linear y DSAP. Cuando la forma homocigota de la enfermedad ocurre en la piel, las manifestaciones pueden ser más tempranas y más severas, con más hiperqueratosis y más lamelas coneoides, como se observó en las lesiones verrucosas del monte de venus y glúteos de ésta paciente.
Por lo tanto el trasfondo de la enfermedad es DSAP heterocigota. La mutación somática y pérdida de la heterocigocidad origina varias formas segmentarias, como la poroqueratosis linear, co-existiendo en el mismo individuo con DSAP. Happle ha designado ésta coexistencia de variantes como una manifestación segmentaria tipo II de una condición autosómica dominante. La mayor incidencia de carcinoma de piel en la variante homocigota de la poroqueratosis linear y posible poroqueratosis palmaris y plantaris (que puede ser un mosaico que involucra piel acral) puede ocasionarse por múltiples factores, como inicio temprano, mayor tamaño, predisposición genética subyacente relacionada a pérdida de la heterocigocidad. No hay estudios definitivos que prueben éstas hipótesis.
Las características clínicas y genéticas de las variantes de poroqueratosis se resúmen en la tabla I. Basados en estudios de familias chinas con DSAP, se identificaron al menos 3 loci candidatos: DSAP 1 (SART 3 en el cromosoma 12q24.1-24.2), DSAP 2 (SSH1 y ARPC3 en el cromosoma 15q 25.1-26.1) y DSAP 3 (en el cromosoma 1p31.3-p31.1). Se identificó un locus potencial para poroqueratosis superficial diseminada (en el cromosoma 18p11.3) y uno para la poroqueratosis palmar y plantar diseminada (en el cromosoma 12q24.1-24.2). SART3 (el antígeno del carcinoma de células escamosas reconocido por las célulasT) es una proteina nuclear que se une al ARN que puede inducir células T citotóxicas y tumor específicas. El SART 3 regula la proliferación y transformación de células epiteliales. La SSH1 es una proteína fosfatasa involucrada en la regulación de la dinámica actínica citoesquelética. La ARPC3 (complejo de proteinas relacionadas con la actina) vincula el filamento de actina en el borde de las células epiteliales migratorias. El perfil de la expresión genética de la lamela corneoide en la poroqueratosis de Mibelli comparado con el perfil en epidermis adyacente normal revela una expresión de ARNm similar a la psoriasis. Estos hallazgos se confirman por las inmunotinciones que muestran sobrexpresión cutánea de productos genéticos up regulados K16, S100AB y A9 y 26 proteinas conexina en la lamela corneoide.
Existen múltiples loci potenciales para ésta enfermedad autosómica dominante con una variedad de manifestaciones clínicas, todas similares bajo el microscopio. La lamela corneoide-capas de paraqueratina más una proliferación clonal de queratinocitos- es una característica diagnóstica histopatológica. Los estudios genéticos sugieren un “defecto en la vía” en la que varias mutaciones diferentes en la proliferación y diferenciación de queratinocitos origina el mismo resultado, la poroqueratosis. La expresión clínica e histopatológica en cada paciente probablemente dependa de las mutaciones particulares y de las vías involucradas.
Tabla I. Expresión clínica y genética de las variantes de poroqueratosis.
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.










