PoliarterItis nodosa cutánea

La poliarteritis nodosa clásica fue la primera vasculitis sistémica descripta. En 1866, Kussmaul y Maier caracterizaron a ésta condición fatal que originariamente se denominó “periarteritis nodosa”. En 1903, Ferrari describió la característica inflamación arterial transmural y propuso el término “poliarteritis nodosa” (PAN). En 1931 Lindberg fue el que primero reconoció a la PAN limitada a la piel. Aunque la poliarteritis nodosa cutánea (CPAN) afecta predominantemente a la piel, los hallazgos extracutáneos incluyen fiebre, malestar general, mialgias, artralgias y neuropatía. Mientras que la PAN sistémica se evidencia generalmente primero por los hallazgos cutáneos de CPAN, hay que investigar compromiso multi orgánico en PAN sistémica, particularmente en riñones, corazón e hígado. Esta distinción es esencial.
La poliarteritis nodosa sistémica es rara; y la PAN cutánea más rara aún, la verdadera incidencia es desconocida. Esta vasculitis cutánea afecta todas las edades. Aunque la mayoría de los estudios no demuestran predominio de género entre los pacientes con CPAN, uno grande encontró una relación hombre: mujer de 1: 1.7. El promedio de edad al momento del diagnóstico fue de 43.5 años para pacientes sin úlceras cutáneas y de 47 años para los que presentaban úlceras.
La etiología es desconocida. La poliarteritis nodosa cutánea es la que más probablemente se considera como una enfermedad inmuno mediada por complejos. La inmunofluorescencia directa (DIF) generalmente muestra depósitos de IgM y de C3 en las paredes arteriales afectadas. La prevalencia de un 77% de complejos IgM antifosfatidilserina-protrombina entre pacientes con CPAN arrojó la hipótesis de que la protrombina unida a células endoteliales apoptóticas induce una respuesta inmune, ocasionando el desarrollo de complejos de anticuerpos antifosfatidilserina-protrombina. Estas inmunoglobulinas presuntamente activan la vía clásica del complemento para causar CPAN.
La poliarteritis nodosa cutánea puede reflejar enfermedad subyacente como infección, o uso de medicación. El agente más comúnmente involucrado es el Estreptococo B hemolítico del grupo A. Se ha demostrado una relación entre CPAN e infección por hepatitis B (4 de 9 pacientes). Aunque la hepatitis B rutinariamente causa PAN sistémica, la correlación entre CPAN y el virus no es consistente. Otros encontraron relación con la enfermedad inflamatoria intestinal. Existen reportes de casos que asocian CPAN con infección por hepatitis C, parvovirus B-19 y micobacterium tuberculosis.
La CPAN inducida por minociclina es un fenómeno bien documentado y resuelve al discontinuar la droga.
Manisfestaciones Clínicas:
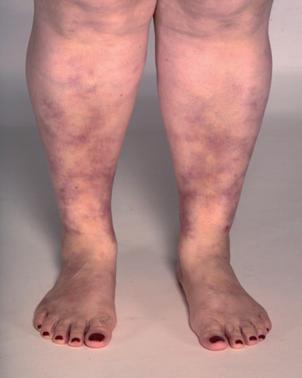
La poliarteritis nodosa cutánea generalmente aparece primero como livedo reticularis (fig 1a), nódulos subcutáneos dolorosos o ulceraciones cutáneas. Otros hallazgos incluyen petequias, púrpuras, necrosis cutánea, autoamputaciones y manifestaciones extracutáneas locales. Esto ocurre más comúnmente en piernas. Las piernas se afectan en el 97% de los casos, seguidas de brazos en el 33% y tronco en el 8%. Se ha notado compromiso de cabeza y cuello en 9 de 23 pacientes (39%) con CPAN.
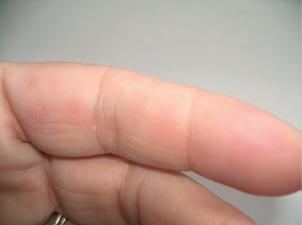
Figura 1 (a) Patrón livedoide extensor en piernas en un caso de poliarteritis nodosa cutánea. (b) Nódulos subcutáneos dolorosos en dedos de una mujer de 57 años.
 a
a
 b
b
Los nódulos pequeños son más palpables que visibles entre los hallazgos más comunes. Estos nódulos, con o sin livedo reticularis, son generalmente la primera manifestación de la enfermedad, y son los precursores de la ulceración en el 50% de los casos. La figura 1b muestra varios de éstos nódulos subcutáneos dolorosos en el dedo de una mujer de 57 años de edad. Un patrón típico de livedo reticularis irregular alrededor de una úlcera es altamente sugestivo de CPAN.
El estudio retrospectivo más extenso hasta la actualidad, analizó a 79 casos de CPAN, encontrando que los nódulos dolorosos en las extremidades inferiores con edema eran los hallazgos más comunes, vistos en el 80% de los casos. El livedo reticulares estaba presente en el 56% de los pacientes, las úlceras cutáneas en el 49% y las placas induradas en el 10%. Otro reporte de 16 pacientes describió la presencia de livedo reticularis en 14 (87.5%) y nódulos subcutáneos en todos los pacientes analizados. Otros encontraron nódulos subcutáneos dolorosos en sólo el 30-50% de los pacientes. El 50% de los pacientes también tenía úlceras cutáneas y el 31.3% tenían lesiones hemorrágicas, desde petequias hasta púrpura extensa.
Las manifestaciones extracutáneas de CPAN incluían síntomas constitucionales, mialgias, artralgias y neuropatía (mononeuropatía y mononeuropatía múltiple).
La base patológica de éstos hallazgos no se han establecido. Los síntomas constitucionales como fiebre se han reportado en el 25-30% de los pacientes. La neuropatía se ha demostrado por electromiografía en el 22% de 79 pacientes con CPAN. El 31.2% de los pacientes con CPAN experimentó mialgias. La miositis y neuritis generalmente resuelve en meses. Las artralgias pueden ocurrir en el 69% de los casos. Las artralgias generalmente son leves, transitorias y ocasionalmente se asocian con artritis no deformante establecida radiológicamente. Sin embargo, pocos pacientes tienen CPAN con artritis severa y progresiva.
La poliarteritis nodosa cutánea tiene un curso benigno crónico con recaídas. Aunque las recurrencias son típicas, las remisiones pueden ocurrir espontáneamente o seguida de la terapia con esteroides. El curso clínico puede durar meses o años, con síntomas agudos o brotes que persisten por 2-8 semanas. Los nódulos subcutáneos pueden tardar años en desaparecer. No se ha demostrado relación entre la edad de inicio y la severidad de la enfermedad en CPAN.
Sin embargo, las autoamputaciones de dedos de manos y pies ocurren en pacientes menores de 10 años de edad. El pronóstico de CPAN es favorable en niños y adultos.
Tal vez el problema de CPAN es la posibilidad de progresión a PAN sistémica. Desafortunadamente, la progresión a PAN sistémica ha sido bien documentada en un estudio donde 2 de 20 pacientes con CPAN desarrollaron PAN sistémica luego de 18 y 19 años de seguimiento.
Diagnóstico:
La poliarteritis nodosa cutánea es una vasculitis de arterias pequeñas y medianas. No existen tests serológicos específicos para CPAN, el diagnóstico requiere de la correlación clínico-patológica. Los característicos nódulos subcutáneos dolorosos en las extremidades inferiores con livedo reticular y/o ulceración cutánea, sugieren la sospecha de CPAN. Pueden evidenciarse manifestaciones extracutáneas como síntomas constitucionales, mialgias, artralgias y parestesias. Se puede obtener una biopsia de piel para llegar al diagnóstico.
Cuando se toma una muestra de un paciente con sospecha de CPAN, es esencial obtener una muestra adecuada. Si está presente la ulceración, la muestra deberá tomarse de una biopsia profunda incisional del borde de la úlcera, en vez de una biopsia por punch. Debería obtenerse una cantidad adecuada de tejido subcutáneo, tejido central a la úlcera, y tejido normal para optimizar el diagnóstico. La razón más común de biopsias repetidas en pacientes con CPAN es la falta de tejido central adecuado al borde de la úlcera. Esto puede acompañarse de una cantidad inadecuada de tejido subcutáneo en la biopsia.
Luego de la confirmación histológica de la presencia de vasculitis involucrando arterias de pequeño y/o mediano calibre, el diagnóstico de CPAN puede hacerse luego de la exclusión de PAN sistémica. Por lo tanto, deben descartarse las manifestaciones sistémicas de PAN. Deben realizarse la medición de la presión arterial, hemograma completo, eritrosedimentación, tests de función hepática y renal, crioglobulinas, y anticuerpos antinucleares (ANA), anticuerpo antineutrofílico citoplasmático (ANCA), factor reumatoideo, y niveles de complemento se indican para excluir otras causas de vasculitis, y distinguir CPAN de PAN sistémica. La evaluación debe basarse en los síntomas específicos. La evaluación de la sintomatología en CPAN incluye electromiograma y enzimas musculares en pacientes con mialgias y debilidad muscular, estudios de conducción nerviosa para los que presentan parestesias, estudio de heces con consideración de angiografía mesentérica y colonoscopía para los pacientes con dolor abdominal y angiograma renal para los pacientes con disfunción renal y/o hipertensión.
El factor causal al inicio de la vasculitis debería pensarse usando títulos ASTO o cultivos de hisopados faríngeos, serología para hepatitis, test de tuberculina y evaluación de condiciones médicas concomitantes como enfermedad inflamatoria intestinal, infección, e historia de medicamentos. El seguimiento debería realizarse 2 veces al año o anualmente en periodos asintomáticos, con incremento de la frecuencia durante las exacerbaciones. En cada evaluación de rutina debe evaluarse para una posible PAN sistémica. Esto comprende una historia y exámen físico completo incluyendo signos vitales, tests de eritrosedimentación, hemograma completo, niveles de complemento, función renal y hepática.
Anormalidades de laboratorio:
Las alteraciones de laboratorio que se encuentran frecuentemente son anemia leve, leucocitosis moderada y eritrosedimentación acelerada. La eritrosedimentación está acelerada en casi el 60% de los pacientes con CPAN. Los títulos de anti-estreptolisina O y/o los cultivos de hisopados farígeos pueden ser de valor para detectar infección estreptocócica, especialmente en niños. En general los tests serológicos para sífilis, ANA, factor reumatoideo y ANCA son negativos. Sin embargo, la positividad ANCA perinuclear se ha encontrado en un caso aislado de CPAN y es evidente frecuentemente en CPAN inducida por minociclina.
Un estudio reciente correlacionó la presencia de anticuerpos asociados con el síndrome antifosfolípido en CPAN y ha encontrado que 13 de 16 pacientes con CPAN (81.3%) tenían positivo el anticuerpo Ig M antifisfatidilserina-protrombina (anti-PS/PT), mientras ningún control fue positivo. Los tres pacientes restantes tenían positivo el anticoagulante lúpico (LAC) o la IgG anti-PS/PT. En comparación con los pacientes con lupus eritematoso sistémico, los pacientes con CPAN tenían niveles significativamente más altos de IgM anti-PS/PT. Un total de 7 pacientes (43.8%) con CPAN eran positivos para LAC. En los pacientes LAC positivos con CPAN, se encontró una correlación significativa entre IgM anti-PS/PT y proteína C reactiva.
Histopatología:
La biopsia de piel profunda incisional, incluyendo tejido subcutáneo revela la presencia de vasculitis leucocitoclásica en las arterias de pequeño y mediano tamaño de la dermis profunda e hipodermis (fig 2). Se han descripto 4 estadíos de hallazgos histológicos en CPAN: degenerativo, inflamatorio agudo, tejido de granulación, y estadío de curación. El estadío degenerativo se caracteriza por degradación de la pared arterial con depósitos de material fibrinoide y destrucción parcial o completa de la lámina elástica interna y externa. El estadío inflamatorio agudo comprende principalmente un infiltrado neutrofílico con eosinófilos, alrededor y en la pared arterial. En el estadío de granulación, éste infiltrado está compuesto incremento de linfocitos y macrófagos, con proliferación intimal y trombosis de la arteria conduciendo a la ulceración. El estadío final de curación muestra una proliferación fibroblástica extendiéndose perivascularmente.
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.










