Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica

Introducción
El Síndrome de Stevens-Johnson (SSJ) y la Necrólisis Epidérmica Tóxica (NET) son reacciones graves aunque poco frecuentes mediadas por células, con extensa necrosis y desprendimiento de la epidermis y complicaciones mucocutáneas en ~ 90% de casos.
El SSJ y la NET sólo se diferencian a lo largo de un espectro de gravedad en base al porcentaje de superficie corporal involucrada (<10 % en el SSJ, 10% a 30% en la superposición SSJ/NET, 30% en la NET). Hay de 1 a 7 y de 0,4 a 1,5 casos por millón de personas por año para el SSJ y la NET, respectivamente, con una incidencia aproximadamente igual entre niños y niñas.
La fisiopatología precisa aún no está clara, pero se cree que el daño de la piel resulta de reacciones citotóxicas mediadas por células y en general fármaco-específicas contra los queratinocitos.
Con sus manifestaciones inespecíficas iniciales y sus altas tasas de mortalidad, estas afecciones representan un desafío diagnóstico significativo para los médicos de emergencias. Aunque la evidencia actual permanece sin pruebas concluyentes sobre posibles medidas curativas, hay pasos críticos que se pueden considerar para disminuir la mortalidad y la morbilidad.
Etiología
La NET es provocada por medicamentos o infecciones respiratorias altas en el 74% a 94% de los casos. En los niños, los medicamentos son los precipitantes más comunes de SSJ/NET. Los medicamentos más frecuentemente involucrados son sulfonamidas, fenobarbital, lamotrigina, carbamazepina, y antiinflamatorios no esteroides (AINEs).
Por lo general, los efectos adversos se presentan dentro de las primeras 8 semanas después de su inicio, con un mayor riesgo a mayores dosis y ante su rápida introducción. En ciertas poblaciones de estudio, existe una asociación genética entre los alelos HLA-B y el SSJ inducido por lamotrigina, fenitoína, carbamazepina y medicamentos fríos.
Los fármacos potencialmente agresivos pueden ser apuntados en base a los seis parámetros de Alden (algoritmo de causalidad de medicamentos para la necrólisis epidérmica): demora de tiempo desde la administración del fármaco al inicio de la reacción, probabilidad de presencia del fármaco en el cuerpo, exposición previa al mismo fármaco independientemente de la reacción en ese momento, presencia del fármaco más allá de la fase de progresión, notoriedad del fármaco como causa de SSJ/NET, y presencia o ausencia de otras etiologías. Los síntomas de SSJ/NET no son atribuidos claramente a un fármaco en el 20% a 25% de los casos (mayor para los niños).
La infección es la segunda causa precipitante más común. Los principales gérmenes involucrados son Mycoplasma pneumoniae, citomegalovirus, herpes virus y virus de hepatitis A. Otras condiciones predisponentes incluyen el VIH (100 veces mayor riesgo), tumores malignos, lupus eritematoso sistémico, radioterapia, enfermedad vascular del colágeno, luz ultravioleta, enfermedades genéticas, y patologías inmunológicas subyacentes.
Es de destacar que algunos estudios han demostrado la recurrencia de SSJ/NET en los sobrevivientes. En un estudio retrospectivo de 55 casos, 10 niños presentaron recurrencia entre 2 meses y 7 años después del primer episodio, 3 tuvieron múltiples recidivas y 1 falleció. Estos hallazgos pueden sugerir una predisposición genética y una vulnerabilidad de larga duración para SSJ/NET.
Curso clínico
Las manifestaciones iniciales son comúnmente fiebre y síntomas de gripe (malestar general, mialgias, artralgias, disfagia, fotofobia, prurito/ardor conjuntival). El compromiso de la piel y la mucosa surge 1 a 3 días después.
Las máculas con centros purpúricos dan paso a grandes ampollas, vesículas y bullas. El desprendimiento epidérmico con el signo de las bullas de Nikolsky (desprendimiento de la capa superficial de la piel con una ligera presión de frotamiento) se presenta 3 a 5 días más tarde y deja denudadas grandes áreas de la piel.
Estos signos se manifiestan primero en la cara y el tórax, y luego se expanden hacia el exterior de forma simétrica. Las porciones distales de los brazos y las piernas están relativamente a salvo, pero las palmas y las plantas pueden ser un sitio temprano de lesión.
Las zonas afectadas son sensibles al tacto. La re-epitelización comienza 1 semana después del inicio del cuadro y puede tardar hasta 3 semanas, aunque es más rápida en niños.
| Las grandes áreas de lesión pueden causar dolor severo, pérdida masiva de líquidos y proteínas, desequilibrios de electrolitos, sangrado, pérdida de calor por evaporación con la subsiguiente hipotermia, resistencia a la insulina, estado hipercatabólico, infección y bacteriemia, shock hipovolémico con insuficiencia renal, y disfunción orgánica múltiple. |
Es de destacar que la piel denudada predispone al paciente a sobreinfección bacteriana, en general por Staphylococcus aureus y Pseudomonas aeruginosa. La sepsis es la principal causa de muerte en los casos de SSJ/NET.
Las complicaciones oftálmicas también están presentes en hasta un 30% de los niños y adultos sobrevivientes, y ocurren dentro de las 2 a 6 semanas del inicio del fármaco. Estas incluyen conjuntivitis severa con secreción purulenta, edema y eritema de párpados, queratitis supurativa, endoftalmitis, dolor, y fotofobia. Lo más preocupante es la pérdida de visión a largo plazo que se puede producir por inflamación crónica de la córnea.
También pueden observarse patologías pulmonares (neumonía, neumonitis intersticial, síndrome de distrés respiratorio agudo, ventilación mecánica en el 25% de los casos con mayor mortalidad asociada), gastrointestinales (diarrea, melena, ulceraciones del intestino delgado, perforación colónica, intususcepción del intestino delgado, estenosis, constricción, estomatitis), vulvovaginitis, y problemas urológicos (uretritis).
Diagnóstico
El síndrome estafilocócico de la piel escaldada es causado por una cepa específica de Staphylococcus con exotoxinas exfoliativas
Las manifestaciones cutáneas de estas dos afecciones pueden parecerse inicialmente al eritema multiforme (EM), al síndrome estafilocócico de la piel escaldada (SEPE), a la enfermedad de Kawasaki (EK), y a la reacción morbiliforme a un fármaco.
El SSJ y la NET tienen una mayor tasa de morbilidad y mortalidad en comparación con la mayoría de las otras etiologías; por lo tanto, deben ser sumamente considerados en el diagnóstico diferencial del médico de emergencia. Sin embargo, existen signos clínicos sutiles que pueden ayudar a distinguir estas afecciones de la piel.
El eritema multiforme es una erupción que se localiza en piel y mucosas, en comparación con el compromiso sistémico del SSJ y la NET. Las lesiones del EM son lesiones diana localizadas periféricamente con limitado desprendimiento epidérmico. A pesar de las numerosas lesiones, el EM típicamente suele comprometer menos del 10% de la superficie corporal. El compromiso de las mucosas está ausente o limitado a una superficie, en general la boca.
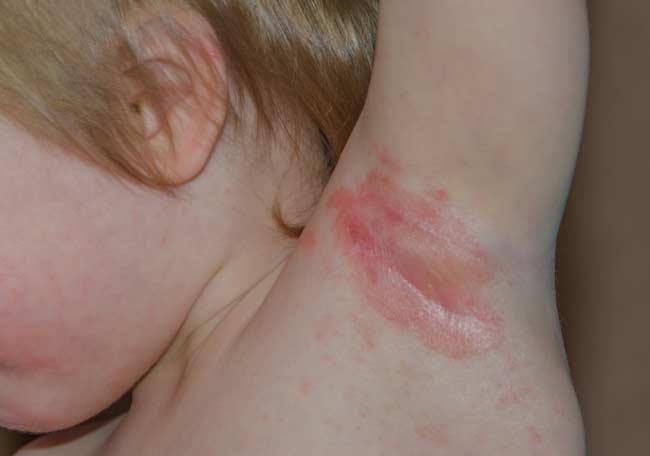
El síndrome estafilocócico de la piel escaldada es causado por una cepa específica de Staphylococcus con exotoxinas exfoliativas. Un pródromo de conjuntivitis purulenta u odinofagia puede estar presente durante 48 horas, seguido por fiebre, malestar general, y hallazgos cutáneos. En niños pequeños, el SEPE puede progresar de un eritema generalizado a la formación de ampollas y descamación, no del todo diferente al SSJ y la NET.
Sin embargo, las lesiones del SEPE nunca son oscuras o purpúricas en apariencia debido a la escisión por la exotoxina de las conexiones mediadas por la desmogleína 1 intracelular en comparación con la necrosis epidérmica del SSJ y el NET.
Las ampollas del SEPE comúnmente afectan las zonas de flexión. En general no hay implicación de las membranas mucosas. Las bullas de ambos síndromes presentan signo de Nikolsky positivo; sin embargo, el SEPE demuestra signo de Nikolsky en la piel no afectada.
Las manifestaciones en piel y mucosas de la EK pueden confundirse con el SSJ y la NET. Ambas entidades pueden presentar fiebre; uno de los criterios clásicos para EK es la fiebre durante al menos 5 días. Típicamente la erupción aparece dentro de los 5 días del inicio de la fiebre y se acompaña a menudo de descamación en las regiones perianal o periungueal.
El exantema polimorfo de la EK se diferencia en que es atípica la presencia de lesiones ampollosas o vesiculares; se describen más comúnmente erupciones morbiliformes, EMs, o eritrodermias. La conjuntivitis bilateral de la EK es no exudativa, en comparación con la conjuntivitis muy exudativa del SSJ, y puede tener preservación límbica patognomónica.
Los cambios en los labios y la cavidad oral incluyen orofaringe eritematosa difusa, labios aframbuesados o rojos y fisurados. Finalmente, los criterios típicos restantes ayudan a diferenciar la EK e incluyen linfadenopatía cervical y cambios en las extremidades tales como el fenómeno de Raynaud.
Las erupciones farmacológicas morbiliformes clásicas, también conocidas como maculopapulares o exantemáticas, pueden confundirse con SSJ y NET incipientes. Las erupciones por drogas son asintomáticas o pruriginosas y más importante aún carecen del compromiso mucoso y del dolor cutáneo característicos.
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.