Mieloma múltiple

El mieloma múltiple es el segundo cáncer hematológico más común (10-15% de todos). Es responsable del 15-20% de las muertes por cáncer hematológico y del 2% de todas las muertes por cáncer. Hoy en día, el mayor conocimiento de su patogénesis ha llevado al desarrollo de nuevos tratamientos.
Aunque el mieloma sigue siendo un cáncer incurable, la supervivencia ha mejorado y los pacientes recién diagnosticados tienen una expectativa de vida de alrededor de 5 años. Con un número cada vez mayor de supervivientes es importante que los profesionales de la salud tengan en cuenta a la enfermedad y los tratamientos actuales.
¿Qué es el mieloma y quiénes lo sufren?
En el mieloma, las células plasmáticas neoplásicas se acumulan en la médula ósea y producen una proteína monoclonal que se detecta en la sangre o en la orina (o en ambas), lo que provoca el deterioro del órgano o de los tejidos. Los estudios epidemiológicos indican que está precedido de una gammapatía monoclonal de significado indeterminado (GMSI), una condición asintomática.
Generalmente afecta a las personas de edad avanzada (edad media al momento del diagnóstico 70 años), aunque el diagnóstico debe ser considerado a cualquier edad porque el 15% de los casos es diagnosticado en las personas <60 años y el 2% en <40 años.
Es 2 veces más común en los afrocaribeños y los sujetos de raza blanca; en todos los grupos raciales la incidencia es 50% mayor en los hombres que en las mujeres. No se conocen componentes genéticos hereditarios o factores de riesgo ambiental definidos.
¿Cuál es la fisiopatología subyacente?
El mieloma se produce debido a los cambios genéticos que tienen lugar durante la diferenciación terminal de los linfocitos B en células plasmáticas. En casi la mitad de los casos se produce una translocación cromosómica por la cual se coloca un oncogén en el gen de la cadena pesada de la inmunoglobulina en el cromosoma 14 (translocación IgH). Esto da como resultado la sobreexpresión del oncogén y la desregulación de la proliferación celular. Los casos restantes se caracterizan por trisomías de varios cromosomas impares, es decir, los cromosomas 3, 5, 7, 9, 11, 15, 19, y 21.
La presencia de estas numerosas trisomías se llama hiperdiploidía. A medida que el mieloma se desarrolla se producen más eventos genéticos, como las mutaciones RAS. Debido a que el crecimiento y la supervivencia de las células del mieloma dependen de otras células de la médula ósea—fibroblastos, osteoblastos, osteoclastos, células del estroma y células dendríticas—se han desarrollado tratamientos que tienen como objetivo el ambiente celular de la médula ósea.
¿Cuál es la causa de la enfermedad de los huesos y de la hipercalcemia en el mieloma?
El desequilibrio de la remodelación ósea en la médula ósea del mieloma está causado por el aumento de la actividad de los osteoclastos y la disminución de la función de los osteoblastos. Las células del mieloma provocan un aumento de la producción de los factores activadores de los osteoclastos y de las citocinas que inhiben la diferenciación de los osteoblastos. La hipercalcemia es producto de la osteólisis sin oposición.
¿Qué causa la insuficiencia renal?
En la mayoría de los casos, las células plasmáticas malignas producen una paraproteína—una inmunoglobulina monoclonal—que en general es la IgG o la IgA. En general, en el mieloma no se encuentra la paraproteína IgM y su presencia sugiere un diagnóstico alternativo, como la macroglobulinemia de Waldenström.
Las células plasmáticas producen también cantidades variables de cadenas livianas monoclonales libres. Las cadenas livianas en la orina, conocidas como proteína de Bence-Jones, se encuentran tanto en el mieloma como en la GMSI. Casi el 20% de los pacientes con mieloma produce solo cadenas livianas en el suero y la orina, mientras que el 2% no produce ni cadenas livianas ni paraproteína; estos pacientes se denominan no-secretores.
Las cadenas livianas se filtran en los glomérulos y se reabsorben en los túbulos proximales. Cuando la carga de cadenas livianas supera esta capacidad de reabsorción, las mismas se precipitan como cilindros en el túbulo distal, causando obstrucción tubular e inflamación tubulointersticial y daño renal agudo.
La nefropatía con cilindros es responsable del 90% de los cuadros de insuficiencia renal relacionada con el mieloma. Otras causas son la deposición de amiloide, la deshidratación, la hipercalcemia, la hiperviscosidad y los fármacos nefrotóxicos, como los antiinflamatorios no esteroides.
¿Cómo se presenta el mieloma?
Las presentaciones más comunes son los síntomas de anemia (observados en el 75% de los pacientes en el momento del diagnóstico), la hipercalcemia (30%), el deterioro renal (25%) y la enfermedad ósea (70%). Las manifestaciones óseas pueden presentarse como lesiones líticas dolorosas, fracturas vertebrales por aplastamiento o fracturas de los huesos largos.
En el 5% de los pacientes con mieloma se producen lesiones de la médula espinal por compresión secundaria a la retropulsión vertebral provocada por las fracturas vertebrales patológicas o por el desarrollo de plasmocitomas en el tejido blando extramedular.
La hipercalcemia, la insuficiencia renal aguda y la compresión medular constituyen emergencias médicas, y el diagnóstico y el tratamiento oportunos son vitales para minimizar el daño a largo plazo de los órganos. La hiperparaproteinemia puede causar síntomas de hiperviscosidad (cefaleas, epistaxis, visión borrosa, y confusión), mientras que la disminución de la inmunidad humoral favorece las infecciones bacterianas recurrentes.
El 30% de los casos es diagnosticado después de un hallazgo incidental, como le elevación de la velocidad de eritrosedimentación, aumento de las proteínas totales o de las inmunoglobulinas. Los síntomas de la presentación suelen ser inespecíficos, como el letargo o el dolor de espalda, lo que puede retrasar el diagnóstico.
En un informe reciente, el 56% de los pacientes que se presenta a la práctica general esperó más de 6 meses para consultar a un hematólogo. Un tercio de los casos se diagnostica en la consulta de urgencia y no por ser casos derivados al hematólogo por el médico general; estos pacientes tienen mal pronóstico (supervivencia al año, 51% vs. 82%).
¿Cómo se diagnostica el mieloma?
En el siguiente cuadro se resumen los criterios diagnósticos para el mieloma, el mieloma asintomático y la GMSI. Esta última se diagnostica cuando la infiltración de las células plasmáticas y las concentraciones de paraproteínas son bajas y el paciente no tiene evidencia de mieloma, como es la hipercalcemia, la insuficiencia renal, la anemia, o las lesiones óseas.
Los pacientes con GMSI tienen alrededor de un 1% de probabilidad anual de progresar al mieloma. Cuando las células plasmáticas o las concentraciones de proteínas monoclonales están elevadas pero no hay ningún órgano o tejido afectado por el mieloma, la condición se conoce como mieloma asintomático. Esta condición tiene un 1% de posibilidad anual de progresar al mieloma sintomático. Un aumento policlonal de las inmunoglobulinas refleja una respuesta inflamatoria aguda y no un GMSI o un mieloma.
|
Criterios del International Myeloma Working Group para el diagnóstico de mieloma Mieloma sintomático Se necesitan 3 de los siguientes diagnósticos: - Células plasmáticas monoclonales en la medula ósea ≥10%. Mieloma asintomático Ambos criterios son necesarios para el diagnóstico: - Proteína monoclonal ≥30 g/L o células plasmáticas monoclonales en la médula ≥10% GMSI Se necesitan los 3 criterios para el diagnostico: - Proteína monoclonal <30 g/L. |
El siguiente cuadro resume las investigaciones que deben realizarse cuando se sospecha un mieloma, destacando las pruebas de detección para los médicos generales.
|
Investigaciones para el diagnóstico de mieloma Pruebas de detección
Pruebas para establecer el diagnóstico
Pruebas para calcular la carga tumoral y el pronóstico
|
La sospecha de mieloma junto con una o más de las siguientes manifestaciones, anemia, insuficiencia renal, hipercalcemia, lesiones líticas en la radiografía o detección de una paraproteína o proteína urinaria de Bence-Jones, debe motivar la derivación a un hematólogo.
Para determinar el grado del mieloma óseo se hará un estudio del esqueleto con radiografías simples de la columna vertebral, el cráneo, el tórax, la pelvis y los huesos largos de los miembros inferiores, y si es necesario, una resonancia magnética, que es la técnica de imagen estándar de oro para investigar la enfermedad vertebral y la posible compresión de la médula espinal. Si no está disponible, se debe realizar una tomografía computarizada.
La gammagrafía ósea no tiene ningún papel en el mieloma porque la captación ósea del tecnecio se basa en la reacción osteoblástica, que en el mieloma es escasa o inexistente. De modo que en la gammagrafía ósea las lesiones líticas del mieloma suelen ser "frías".
La tomografía computarizada por emisión de positrones puede ser importante para el cribado y el control de las localizaciones extramedulares de la enfermedad, especialmente si el mieloma es no secretor.
¿Cuáles son los factores pronósticos?
A pesar del desarrollo de nuevos agentes terapéuticos que transformaron el panorama para muchos pacientes, el mieloma sigue siendo una enfermedad heterogénea. Algunos pacientes viven más de 8 años después del diagnóstico, mientras que un subgrupo de alto riesgo morirá en 24 meses. El sistema de estadificación internacional define 3 categorías de riesgo basadas en las concentraciones séricas de la microglobulina β2 y la albúmina.
|
Sistema de estadificación internacional Estadio I Estadio II Estadio III |
Las lesiones genéticas específicas o las firmas genéticas se asocian con mala evolución. Las translocaciones IgH afectan a los cromosomas 4 y 16, denominadas t(4;14) y t(14;16), las que se consideran de alto riesgo y están asociadas a un mal pronóstico.
El gen supresor de tumores P53 está localizado en el brazo corto, o brazo p, del cromosoma 17, y la supresión de este brazo (del 17p) también se asocia con un mal resultado. Los pacientes con translocaciones de IgH t(11;14) o t(6;14) se consideran con riesgo normal, al igual que los pacientes con hiperdiploidía.
La edad es un factor pronóstico independiente y depende de cómo responde el paciente al tratamiento. El logro de una respuesta completa se asocia a la supervivencia libre de progresión y a la supervivencia global prolongada.
Los pacientes más jóvenes cuyo estado general permite usar dosis elevadas tienen ahora una mediana de la proyección de la supervivencia de alrededor de 7 años.
Los pacientes que se presentan con un cuadro de emergencia tienen peor pronóstico, y cuando hay un retraso diagnóstico superior a los 6 meses existe tendencia a una menor supervivencia global, como lo mostró un pequeño estudio retrospectivo de revisión de casos.
¿Cómo se trata el mieloma?
En la práctica, los pacientes con GMSI y mieloma asintomático no se tratan hasta que se desarrolla el mieloma sintomático. No se ha hallado ninguna intervención para retrasar o prevenir la progresión del GMSI a mieloma. Los pacientes con mieloma asintomático deben ser objeto de seguimiento bajo la supervisión de un hematólogo.
Ensayos controlados aleatorizados hallaron que en el mieloma asintomático la quimioterapia no modificó la supervivencia. Se debe ofrecer a los pacientes participar de las investigaciones sobre el uso de nuevos agentes terapéuticos para las personas con alto riesgo de progresión al mieloma sintomático.
El British Committee of Standars in Haematology (BCSH) recomienda que los pacientes con GMSI y bajo riesgo de progresión al mieloma sean monitoreados en atención primaria, mientras que las personas con alto riesgo de progresión deben ser supervisadas por un especialista.
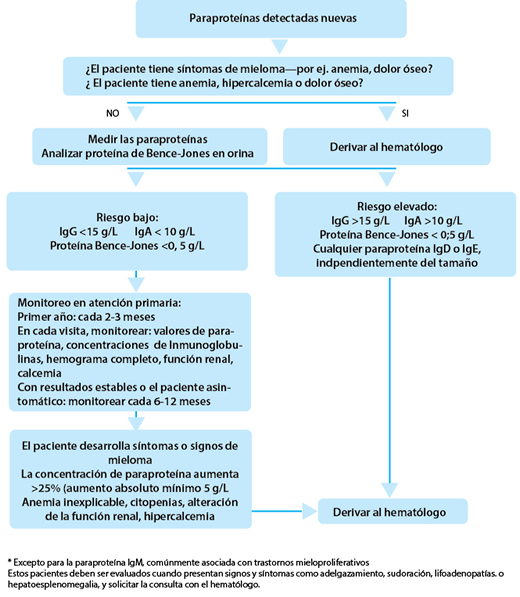
En la última década se ha producido un avance sin precedentes en el tratamiento del mieloma sintomático debido a la introducción del bortezomib (un inhibidor del proteosoma), la talidomida y la lenalidomida, que son fármacos inmunomoduladores.
En la actualidad, estos agentes son la base del tratamiento. La mayoría de los pacientes responde al tratamiento inicial y entra en un período de estabilidad de la enfermedad que se asocia generalmente con una buena calidad de vida.
Debido a la falta de un tratamiento curativo, la recaída es inevitable, pero al menos en un segundo momento, la mitad de los pacientes responde a la quimioterapia con fármacos semejantes o diferentes. Las recaídas posteriores se vuelven cada vez menos sensibles al tratamiento y la enfermedad entra en la etapa final hasta que se hace refractaria, a veces con manifestaciones extramedulares y citopenias.
El enfoque para el tratamiento sintomático del mieloma de reciente diagnóstico depende de la edad y de las comorbilidades. Los regímenes de quimioterapia al iniciar el tratamiento tienen como objetivo conseguir la mayor respuesta con la menor toxicidad, y para los pacientes jóvenes (generalmente <65 años) y con buen estado general, esto se consolida con la quimioterapia a dosis elevadas y con el trasplante autólogo de células madre.
Los pacientes mayores o aquellos con graves comorbilidades que no están en buen estado general para someterse al trasplante autólogo se tratan con quimioterapia sola. La respuesta al tratamiento se clasifica de acuerdo con la reducción de las paraproteínas o de las cadenas livianas.
|
Clasificación de la respuesta de la enfermedad o la progresión al mieloma Tradicionalmente esto se hace sobre la base de la magnitud de la reducción (o el aumento) de las paraproteínas, pero también se tiene en cuenta el grado de plasmocitosis en la médula ósea, la progresión de las lesiones óseas y la existencia de plasmocitomas en los tejidos blandos. Debido al perfeccionamiento de las técnicas para la detección de células del mieloma residuales en la médula ósea se han identificado nuevas profundidades de respuesta, como la respuesta completa estricta. Respuesta completa No se detecta la paraproteína y desaparecen cualquier plasmocitoma de los tejidos blandos y las células plasmáticas en la médula ósea son <5%. Las paraproteínas se reducen en más del 90% o las paraproteínas son detectables pero en cantidades demasiado baja como para ser cuantificadas. Respuesta parcial Reducción de las paraproteínas en más del 50%. No cumple con los criterios de respuesta de la enfermedad o la progresión. Progresión de la enfermedad Hay por lo menos un aumento del 25% en las paraproteínas (aumento de al menos 5 g/L), se desarrollan lesiones óseas nuevas o plasmocitomas, o hay hipercalcemia (calcio sérico corregido>2,65 mmol/l). |
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.