La resolución del caso: ¿Cuál es su diagnóstico? IX

SEMINARIO ANÁTOMO-CLÍNICO 1º CÁTEDRA DE CLÍNICA MÉDICA Y TERAPÉUTICA - CÁTEDRA DE ANATOMÍA PATOLÓGICA FACULTAD DE CIENCIAS MÉDICAS. UNIVERSIDAD NACIONAL DE ROSARIO (Santa Fe, Argentina) Sitio web: www.clinica-unr.org
![]()
Servicios de Clínica Médica y Anatomía Patológica del Hospital Provincial del Centenario. Rosario Correspondencia: lparodi@clinica-unr.org Editores: Dr. Roberto Parodi – Dra. Natalia Egri – Dr. Ramón Ferro - .Dr. Damián Carlson - Prof. Dr. Alcides Greca
Duración: 1 minutos
|
* Si tiene problemas para visualizar este video actualice aquí su Windows Media Player |
 |
Discusión del caso clínico
Dra. Silvina Lema
Se presenta el caso de una mujer de 57 años, con antecedentes de insuficiencia renal crónica en hemodiálisis desde hace 7 años de causa no filiada, con varias internaciones por cuadros que fueron interpretados cómo neumonías agudas de la comunidad, con laboratorio inmunológico positivo para p-ANCA, FAN e hipocomplementemia, estudios de imágenes compatibles con ocupación alveolar y una broncofibroscopia que informa sangrado.
Comenzaré planteando las posibles aproximaciones diagnósticas de la enfermedad de base, y por último analizaré la causa de su deceso.
El cuadro de presentación en esta internación de tos, expectoración hemoptoica, disnea, fiebre, hipoxemia y caída del hematocrito, con aumento de los infiltrados pulmonares en las radiografías de tórax, la rápida progresión en horas y los antecedentes mencionados, me permiten interpretar el caso como un síndrome de hemorragia alveolar difusa (HAD).
Este síndrome puede ser definido por la presencia de hemoptisis, infiltrados alveolares difusos y caída del hematocrito. Sin embargo, más de un tercio de los pacientes no tienen hemoptisis, los infiltrados alveolares pueden ser unilaterales y la caída del hematocrito o hemoglobina pueden ser dificultosos de documentar. El sangrado en los espacios alveolares caracteriza a este síndrome de HAD. Histológicamente puede observarse uno de los tres patrones de lesión:
► Capilaritis pulmonar: está dada por la infiltración neutrofílica de los septos alveolares, produciendo necrosis de dichas estructuras. Esto lleva a una pérdida paulatina de la integridad estructural capilar, permitiendo de ésta manera la extravasación de los glóbulos rojos al espacio alveolar y al intersticio pulmonar. La mayor parte de los neutrófilos, al fragmentarse, liberan radicales libres de oxígeno y enzimas proteolíticas a los espacios alveolares e intersticio, produciendo injuria pulmonar. Los restos celulares de los neutrófilos picnóticos y fibrina ingresan también a los espacios alveolares, produciendo necrosis fibrinoidea.
► Hemorragia alveolar leve: consiste en la hemorragia dentro de los espacios alveolares, sin evidencia de inflamación o destrucción de las estructuras alveolares y de la membrana basal.
►Daño alveolar difuso: es la lesión típica del distrés respiratorio del adulto (SDRA), consistiendo en edema de los septos interlobulillares y presencia de membranas hialinas a lo largo de los espacios alveolares. Este tipo de lesión puede dar sangrado pulmonar.
Con capilaritis pulmonar
♦ Vasculitis primarias idiopáticas de pequeños vasos
Granulomatosis de Wegener
Síndrome de Churg-Strauss
Poliangeítis microscópica
♦ Vasculitis primaria por inmunocomplejos (IC)
Síndrome Goodpasture
Púrpura Schönlein-Henoch
♦ Vasculitis secundarias
Lupus eritematoso sistémico
Artritis reumatoidea
Síndrome antifosfolipídico
Enfermedad mixta del tejido conectivo
Polimiositis/dermatomiositis
Crioglobulinemia esencial
Enfermedad de Behcet
Transplante de pulmón o médula ósea
Inducido por drogas
Sin capilaritis pulmonar (Hemorragia alveolar leve)
♦ Hemosiderosis pulmonar idiopática
♦ Coagulopatía
♦ Estenosis Mitral
♦ Injuria por inhalación
♦ Síndrome Goodpasture
♦ Lupus eritematoso sistémico
♦ Transplante de médula ósea
La presentación clínica puede tener una constelación de síntomas, signos y resultados de laboratorio que pueden sugerir un diagnóstico específico o solamente permite el diagnóstico general del síndrome sin orientar hacia una etiología en particular.
Los síntomas son de inicio brusco y de corta duración, menos de 7 días. Tos, fiebre y disnea son comúnmente las manifestaciones iniciales. Este cuadro puede resolverse en pocos días. Algunos pacientes, sin embargo pueden presentarse con un distrés respiratorio agudo que requiera asistencia respiratoria mecánica. La hemoptisis puede estar ausente en más del 33% de los casos con HAD de cualquier causa. En el caso de nuestra paciente, la presencia de nuevos infiltrados alveolares difusos, una caída leve de la hemoglobina y hallazgos de fluido hemorrágico en el lavado bronquioloalveolar favorecen el diagnóstico.
Los hallazgos radiográficos consisten en nuevas opacidades alveolares difusas, en la fase aguda de la hemorragia, usualmente bilaterales, respetando los ápices y la periferia del pulmón. Son indistinguibles del edema pulmonar en infecciones difusas. Luego de dos o tres días la sangre depositada en los alvéolos es absorbida por el intersticio dando lugar a un patrón reticular. Si el sangrado no recurre la radiografía de tórax se normaliza en una o dos semanas. Episodios recurrentes de HAD pueden llevar a fibrosis pulmonar y opacidades intersticiales.
Cómo anormalidades de laboratorio podemos encontrar una velocidad de eritrosedimentación elevada, leucocitosis, caída del hematocrito e insuficiencia renal en caso de síndrome pulmón-riñón.
En los test de función pulmonar podemos encontrar grados variables de hipoxemia, siendo un marcador sensible para HAD el incremento en la capacidad de difusión de monóxido de carbono (DLCO).
El lavado bronquioloalveolar constituye una modalidad diagnóstica sumamente útil para los pacientes con infiltrados pulmonares difusos sin hemoptisis. Realizado en forma secuencial podría mostrar una hemorragia progresiva en pacientes con HAD, siendo característico el hallazgo de macrófagos cargados de hemosiderina.
Ahora bien, como las causas para HAD son numerosas, haré referencia sólo a las que estimo que más se acercan al posible diagnóstico de este caso.
A pesar de su baja incidencia en el LES, existen comunicaciones en la literatura que avalan las características presentes en este caso, así por ejemplo, en una revisión sobre HAD en LES de Cleveland, describieron su experiencia con 8 pacientes con LES que fueron admitidos por HAD; concluyeron que la HAD podría simular una neumonía y la hemoptisis podría no ser evidente.
Un interrogante que surge es qué relación tiene el p-ANCA anti-mieloperoxidasa que se detectó en varias oportunidades en el contexto de un probable LES. A pesar de que los anticuerpos anticitoplasma de neutrófilos fueron primero asociados a vasculitis primarias, se sabe que un 15-20% de los pacientes con lupus tiene ANCA positivo y su mayor relación es con el patrón peri-nuclear. Además el ANCA anti-mieloperoxidasa presenta asociación con el lupus inducido por drogas. Los pacientes con LES y presencia de p-ANCA en el laboratorio inmunológico tienen más riesgo de desarrollar hemorragia pulmonar por lo que requieren un seguimiento más estricto. Además se debe tener en cuenta que los ANCA también se pueden presentar en otras entidades como artritis reumatoidea, síndrome de Sjögren, miopatías inflamatorias, artritis crónica juvenil, artritis reactivas, policondritis recidivante, esclerodermia y síndrome antifosfolipídico, con un patrón peri-nuclear más que central.
Son también conocidas como vasculitis asociadas a ANCA e incluyen a la granulomatosis de Wegener, el síndrome de Churg-Strauss, la poliangeítis microscópica y dentro de esta última como afectación de órgano específico, la capilaritis pulmonar pauci-inmune aislada y la glomerulonefritis rápidamente progresiva (GNRP) pauci-inmune idiopática. Estas vasculitis son agrupadas ya que presentan características clínicas comunes, afectación patológica de pequeños vasos, respuesta similar a inmunosupresores y ANCA positivo.
El análisis de la sensibilidad, especificidad y el valor predictivo positivo (VPP) del c-ANCA (o anti-proteinasa 3) para la granulomatosis de Wegener y de p-ANCA (o anti-mieloperoxidasa) para el síndrome de Chrug-Strauss, poliangeítis microscópica y la GNRP pauci-inmune idiopática, son importantes en la determinación de su utilidad diagnóstica. c-ANCA es altamente sensible (S 90–95%) en la granulomatosis de Wegener activa generalizada con una especificidad aproximadamente del 90%. En un contexto clínico adecuado, con una alta probabilidad pretest de enfermedad, un c-ANCA anti-proteinasa 3 positivo tiene un valor predictivo positivo suficiente cómo para diferir la biopsia pulmonar. Por otro lado, un p-ANCA positivo no tiene especificidad suficiente y sólo proporciona evidencias sugestivas de vasculitis primarias de pequeños vasos, ya que éste puede ser hallado en una amplia variedad de enfermedades, incluidas la AR y el síndrome de Goodpasture. Por lo tanto, al no presentar nuestra paciente un c-ANCA, alejo la granulomatosis de Wegener como posible diagnóstico y haré referencia entonces a la poliangeítis microscópica.
Estos anticuerpos se pueden demostrar en tejidos a través de la inmunofluorescencia la cuál evidencia los depósitos lineales de IgG dentro del glomérulo y/o en las paredes capilares alveolares. La mayoría presenta síntomas respiratorios con evidencia de enfermedad renal. En un 60-80% existe compromiso renal y pulmonar simultáneo, en un 30% sólo enfermedad renal y menos del 10% enfermedad limitada al pulmón. El diagnóstico se basa en las características clínicas y el hallazgo de anticuerpos circulantes o en los tejidos contra las membranas basales, siendo la sensibilidad del 97% y la especificidad del 98%. El patrón de oro lo constituye la biopsia, mediante la demostración de los depósitos lineales en la membrana glomerular o alveolar. A pesar de presentar afectación renal y pulmonar, nuestra paciente tiene anticuerpos negativos, por lo cuál lo considero un diagnóstico alejado.
El síndrome de dificultad respiratoria aguda del adulto (SDRA), desorden que aparece al final del espectro de “injuria pulmonar aguda” ha sido descripto en pacientes con LES. Por ejemplo en un estudio retrospectivo de 544 pacientes con LES, 19 desarrollaron SDRA, más comúnmente con bacteriemia o sepsis por bacilos Gram negativos. El SDRA fue peor en aquellos que habían sido tratados con glucocorticoides en el mes previo y estuvo asociado a una pobre sobrevida (68 % mortalidad).
En esta paciente, con un estado de inmunosupresión grave, insuficiencia respiratoria aguda e infiltrados bilaterales difusos, puedo plantear como causa para dicho cuadro un SDRA por sepsis.
Dres. Eliana Scaramuzza y Analía Nocito
DIAGNÓSTICOS FINALES:
1. ENFERMEDAD PULMONAR VINCULABLE A ETIOLOGÍA INMUNOLÓGICA: Poliangeítis microscópica.
2. ENFERMEDAD RENAL EN ESTADÍO TERMINAL DE ETIOLOGÍA NO DILUCIDADA: Poliangeítis Microscópica
3. QUISTES RENALES SECUNDARIOS A DIÁLISIS PROLONGADA.
4. BRONCONEUMONÍA MULTIFOCAL BILATERAL.
5. NÓDULO CALCIFICADO EN CARA ANTERIOR DEL LÓBULO INFERIOR IZQUIERDO, CON ADHERENCIAS PLEURO–PLEURALES, ASOCIADO A MÚLTIPLES ADENOPATÍAS CALCIFICADAS: ¿Tuberculosis?
6. CONGESTIÓN HEPATO ESPLENICA
7. DIVERTICULOSIS COLÓNICA.
8. COLELITIASIS.
El caso corresponde a una paciente de 57 años, en diálisis trisemanal por insuficiencia renal de larga data de causa no filiada. Padecía, además, una enfermedad pulmonar crónica de aproximadamente 5 años de evolución, caracterizada por episodios reiterados de disnea, tos con expectoración hemoptoica y sensación subjetiva de fiebre, por los cuales requirió múltiples internaciones.
Teniendo en cuenta el cuadro clínico, sumado a los resultados obtenidos del laboratorio inmunológico, se consideró que la paciente era portadora de una patología autoinmune por lo que fue tratada con corticoides sistémicos.
En su última internación presentó severo compromiso respiratorio, asociado a importante caída del hematocrito, por lo que se decidió su ingreso a unidad de terapia intensiva, donde falleció al día siguiente.
El examen necrópsico reveló que la paciente era portadora de una patología crónica que afectaba fundamentalmente a pulmones y riñones.
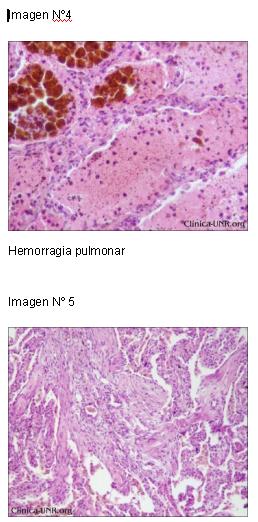
A nivel pulmonar, el hallazgo cardinal fue la hemorragia de carácter reciente y antiguo, que ocupaba las luces alveolares y se asociaba a focos de bronconeumonía bilateral, edema y fibrosis intersticial.
Comentarios
Para ver los comentarios de sus colegas o para expresar su opinión debe ingresar con su cuenta de IntraMed.






